
Rakesh Tiwle
Rungta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Ajazuddin
Rungta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Tapan Kumar Giri
Rungta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Dulal Krishna Tripathi
Rungta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Vishal Jain
University Institute of Pharmacy, Pt. Ravishankar Shukla University, Raipur, Chhattisgarh, India
Amit Alexander
Rungta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Trends in Applied Sciences Research
Year: 2012 | Volume: 7 | Issue: 8 | Page No.: 596-619







ABSTRACT
The combinatorial chemistry and high throughput screening increases the solubility of poorly water soluble compounds. The most challenging task in development of a formulation is the solubility of drug, availability at the site of action and stability of drug. Aqueous solubility of any therapeutically active substance is a key property as it governs dissolution, absorption and thus the in vivo efficacy. Among all newly discovered chemical entities about 40% drugs are lipophilic and these drugs are rejected by the pharmaceutical industry and will never benefit a patient because of its poor bioavailability due to low water solubility and/or cell membrane permeability. Drug efficacy can be severely limited by poor aqueous solubility and some drugs also show side effects due to their poor solubility. Therefore, drug release profiles are exhibited by such formulations for poorly soluble drugs to improve the solubility of such poorly soluble drugs. Any drug to be absorbed must be present in the form of an aqueous solution at the site of absorption. Water is the solvent of choice for liquid pharmaceutical formulations. Most of drugs which are weakly acidic and basic show poor aqueous solubility hence various methods like, salt formation, co-solvency, micronization, addition of agent, solid dispersion, complexation etc., are some of the vital approaches routinely employed to enhance the solubility of poorly soluble drugs. This article reviews various methods used for improving the solubility of hydrophobic drugs and improve the drug release profiles which are exhibited by such formulations for poorly soluble drugs.
PDF Abstract XML References Citation
Received: March 27, 2012;
Accepted: April 23, 2012;
Published: July 24, 2012
How to cite this article
Rakesh Tiwle, Ajazuddin, Tapan Kumar Giri, Dulal Krishna Tripathi, Vishal Jain and Amit Alexander, 2012. An Exhaustive Review on Solubility Enhancement for Hydrophobic Compounds by Possible Applications of Novel Techniques. Trends in Applied Sciences Research, 7: 596-619.
URL: https://scialert.net/abstract/?doi=tasr.2012.596.619
URL: https://scialert.net/abstract/?doi=tasr.2012.596.619
INTRODUCTION
The combinatorial screening programs employed by the pharmaceutical companies identified that about 40% of active New Chemical Entities (NCEs) are poorly water soluble. The two major obstacles in developing a therapeutic agent are Solubility and stability (Seedher and Sharma, 2007). Since 1995, more than 90% of drugs are approved as hydrophobic having poor solubility. A maximum amount of solute dissolved in a given solvent at a specified temperature defined as solubility (Patil et al., 2011). The substance which is to be dissolved is known as solute and the fluid (medium) in which the solute to be dissolve is known as solvent and the process of dissolving solute into solvent is called as solution. Descriptive terms for solubility are shown in (Table 1) (Beringer, 2005).
| Table 1: | Solubility definitions (Rodier et al., 2005) |
 | |
| Table 2: | Biopharmaceutical classification system (BCS) (Malpani et al., 2009) |
 | |
The poorly soluble agent have low water solubility hence they low bioavailability and absorption (Heimbach et al., 2007; Nourani et al., 2008; Vahedi, 2012). There are various techniques and formulations have been employed to overcome these limitations. Although, existing strategies such as complexing drugs by using Cyclodextrins (Vyas et al., 2008; Zhixun et al., 2006; Sangshetti et al., 2008) conjugation to dendrimers (Gupta et al., 2006), salt formation of ionizable drugs (Serajuddin, 2007) and the use of co-solvents (Akers, 2002; Strickley, 2004) have been shown to improve drug solubility. The World Health Organization (WHO) have classified BCS classification on the basis of data as 130 orally administer drug from which according to WHO list 61 could be classified as poorly soluble drug (Al Omari et al., 2009) (Table 2). Biopharmaceutical Classification System (BCS) many drugs belongs to Biopharmaceutics Classification System (BCS) class II (high permeability, low solubility) or IV (Low permeability, Low solubility) (Amidon et al., 1995; Porter and Charman, 2001). For the BCS class II drugs, the oral absorption is limited by the solubility or dissolution in gastrointestinal (GI) tract.
Solubilisation process: The breaking of inter-ionic or intermolecular bonds in the solute occurs mainly in the method of solubilisation. In solubilisation method the solvent provide space for the solute, interaction between solvent and the solute molecule or ion (Fig. 1).
FACTORS AFFECTING SOLUBILITY
Polymorphs: Absorption and bioavailability can also be enhanced by polymorphs as defined as the greater the solubility of the metastable form Blagden et al. (2007) and Ajazuddin et al. (2011). Polymorphs can vary in melting point. Since, the melting point of the solid is related to solubility, the capacity for a substance to crystallize in more than one crystalline form is polymorphism. It is possible that all crystals can crystallize in different forms or polymorphs. If the change from one polymorph to another is reversible, the process is called enantiotropy. If the system is monotropic, there is a transition point above the melting points of both polymorphs. So, polymorphs will have different solubility (Worthen, 2006; Noorizadeh and Farmany, 2011).
Particle size: The solubility of crystalline solids gets affected by particle size it is well describe in the documented (Hammond et al., 2007; Wu and Nancollas, 1998; Mosharraf and Nystrom, 1995).
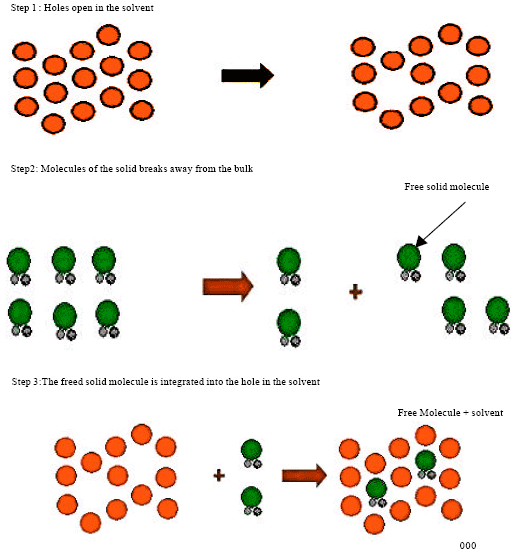 | |
| Fig. 1: | Solubilisation process (Dabbagh and Taghipour, 2007; Sangshetti et al., 2008) |
By reducing the particle size, the solubility of crystalline drugs can be increased to submicron levels, but the effect of solubility is trifling if the particle size is not reduced below 10 μm. The effect of particle size on solubility can be described by Chaumeil (1998):
| Where: | ||
| S | : | The solubility of infinitely large particles |
| So | : | The solubility of fine particles |
| V | : | Molar volume |
| g | : | The surface tension of the solid |
| γ | : | The radius of the fine particle |
Pressures: An increase in pressure increases solubility for gaseous solute. While decreases in pressure for solids and liquid solutes, changes in pressure have practically no effect on its solubility (Ain et al., 2009). There are various approaches to improve the solubility or to increase the available surface area for dissolution. These can be altered or modified by following the methods of Leaner and Dressman (2000).
Temperature: Solubility changes with the temperature. It is demonstrated by Pore and Kuchekar (2011), in solubilisation process energy get absorbs then the temperature will increased and their solubility will increases. If the temperature will increases enhance solubility decrease. A few solid solutes are less soluble in warm solutions (Lindenberg et al., 2004). The solubility of gases deceases with the increasing temperature.
METHOD FOR SOLUBILITY ENHANCEMENT
Physical modifications
Particle size reduction
Micronization: Surface area for dissolution can be increases by Micronization (Kawashima et al., 1975). Micronisation increases the dissolution rate of drug through increased surface area but does not enhanced equilibrium solubility. The increase in bioavailability after micronization of drugs, e.g., by jet or ball milling Example, danazol (Liversidge and Cundy, 1995), progesterone (Hargrove et al., 1989), or dioxin (Jounela et al., 1975).
Nanosuspension: Nanosuspensions are sub-micron colloidal dispersion of pure particles of drug, which are stabilized by the surfactants. (www.expresspharmapulse.com). Nanosuspensions in aqueous or non-aqueous vehicles can be produced by bottom-up (e.g., precipitation) or top-down (e.g., wet milling) processes (Rainbow, 2004; Douroumis and Fahr, 2006). High pressure homogenizers such as the piston gap homogenizer have proved to be a highly successful technology in nanosuspension formation.
Homogenization: Homogenization the required technique is used to reduce the globule size of a coarse emulsion (Amit et al., 2011), globule size is less than 100-200 nm (Davis et al., 1974). Brownian movement prevents creaming because of small globule size which also promotes good physical stability (Floyd, 1999; Chattopadhyay et al., 2011). There are so many method used to improve the dissolution of hydrophobic drugs. High-Pressure Homogenization (HPH) has been mostly used to reduce the particle size (Uchiyama et al., 2011; Grau et al., 2000). For example processing highly concentrated suspensions (Muller et al., 2001) and preparing emulsions (Tian et al., 2007). HPH has lot of advantages over other milling techniques as it is very simple, time saving and an organic solvent-free process. Therefore, HPH can be used to enhance the solubility of hydrophobic drugs such as PLH for which usage of organic solvents is limited (Al-Haj and Rasedee, 2009; Ajazuddin and Saraf, 2010b). This method having some advantages for Production of Solid Lipid Nanoparticles (SLNs) (Bhoyar et al., 2012; Ajazuddin and Saraf, 2010a). The objective of this study was to investigate solid lipid nanoparticles using Carbopol gel as gelling agent containing triamcinolone acetonide acetate (glucocorticoid compound) for transdermal iontophoretic delivery Solid Lipid Nanoparticles (SLN) (Mehnert and Mader, 2001; Muller et al., 2001) have been introduced to the literature as a carrier system for poorly water soluble pharmaceutical drugs (Ugazio et al., 2002; Westesen et al., 1997; Lokhande et al., 2006; Nourani et al., 2008) and cosmetic active ingredients.
Wet milling: Active drug in the presence of surfactant is defragmented by milling (Aulton, 2002). Other technique involves the spraying of a drug solution in a volatile organic solvent into a heated aqueous solution. Rapid solvent evaporation produces drug precipitation in the presence of surfactants.
MODIFICATION OF THE CRYSTAL HABIT (Hite et al., 2003)
Polymorphs: Polymorphism is the ability of compound to crystallize in more than one crystalline form. Different polymorphs of drugs are chemically identical, but they exhibit different physicochemical properties including solubility, melting point, density, texture, stability etc. Generally, the anhydrous form of drug has greater solubility than the hydrates. This is because the hydrates are already in interaction with water and therefore, have less energy for crystal breakup in comparison to the anhydrites (i.e., thermodynamically higher energy state) for further interaction with water (Hammond et al., 2007; Chattopadhyay et al., 2011).
DRUG DISPERSION IN CARRIERS
Solid dispersion technique: The concept of solid dispersions was given by Sekiguchi and Obi (1961) who investigated the generation and dissolution performance of eutectic melts of a sulfonamide drug and a water-soluble carrier in the early 1960s (Tapas et al., 2011; Giri et al., 2010; Zhixun et al., 2006). Many of the drugs belongs to these techniques; can be categorized as class II according to the Biopharmaceutical Classification System (BCS). These drugs are poorly water soluble but once they are dissolved they get easily absorbed through the gastro-intestinal membrane. One of the approaches to enhance the dissolution rate is the use of solid dispersion. Some marketed formulation of solid dispersion shown in (Table 3).
Definition of solid dispersions: The two different components, generally a hydrophilic matrix and a hydrophobic drug mainly consist of solid dispersion (Chiou and Riegelman, 1971). These matrix are either crystalline or amorphous. In both particle (amorphous particles or crystalline particles) the drug can be dispersed molecularly (Ajazuddin et al., 2011). Solid dispersion is describing the most promising method to improve the oral bioavailability of hydrophobic drugs by preparing Lipid Nano Spheres (LNSs) (Amarji et al., 2007). There are different approaches which can be used for increasing the dissolution hydrophobic drugs of t he as given in the figure Fig. 2. That describes the approaches to Increase solubility/Dissolution (Verma, 2011; Patidar et al., 2010).
| Table 3: | Marketed formulation of solid dispersion (Patel et al., 2010) |
 | |
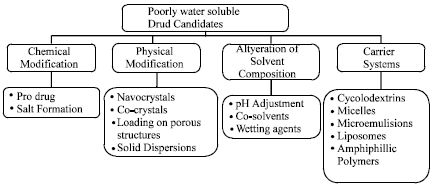 | |
| Fig. 2: | Approaches to increase solubility/dissolution (Verma, 2011) |
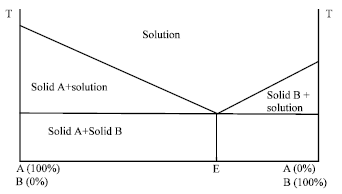 | |
| Fig. 3: | Phase diagram of a simple eutectic mixture with negligible solid solubility, (Sharma et al., 2009).T A: M.P. of solid A (in °C), T B: M.P. of solid B (in °C ), TE: Eutectic point |
Categories of solid dispersions
Simple eutectic mixtures: The two components which are completely miscible in a liquid state but only to a very limited extent in the solid state forms a simple eutectic mixture (Fig. 3) (Sharma et al., 2009). When, a composition E with a mixture of A and B is cooled, at first A and B crystallize out simultaneously, whereas when other compositions are cooled, one of the components starts to crystallize out while after that when composition E is further cooled one component starts to crystallize out before the others (Goldberg et al., 1966). Solid eutectic mixtures are usually prepared by rapid cooling of a co-melt of the two compounds in order to obtain a physical mixture of very fine crystals of the two components. When a mixture with composition E, consisting of a slightly soluble drug and an inert, highly water soluble carrier, is dissolved in an aqueous medium, the carrier will dissolve rapidly, releasing very fine crystals of the drug. Where T A-M.P. of solid A (in °C), T B-M.P. of solid B (in °C), TE-Eutectic Point.
Solid solution: Solid solutions of a poorly water soluble drug dissolved in a carrier with relatively good aqueous solubility are of particular interest as a means of improving oral bioavailability (Leaner and Dressman, 2000). Two components crystallize together in homogenous one phase system. Particle size of drug in solid solution is reduced to its molecular size. Solid solutions shows faster dissolution rate than eutectic mixtures. Solid solutions can be divided in two types, according to their miscibility (continuous versus discontinuous solid solutions) or, according to the way in which the solvate molecules are distributed in the solvendum (substitutional, interstitial or amorphous).
Miscibility types
Continuous: The continuous solid solution consists of totally miscible components both in liquid and solid state (Giri et al., 2010). The pure components in a solid state lattice energy as compare to continuous solid solution it is due to the higher heteromolecular bonding than the homomolecular one in a continuous solid solution (Fig. 4) shows the hypothetical phase diagram of a continuous solid solution.
Discontinuous solid solutions: Discontinuous solid solutions, the miscibility or solubility of one component is restricted in other (Fig. 5) shows a typical phase diagram of a discontinuous solid solution. α and β shows the regions of true solid solutions. The region labeled β is a solid solution of B in A that is component A would be regarded as the solvent and B as the solute. Similarly the region labeled β is a solid solution of A in B (Goldberg et al., 1965).
The way in which the solvate molecules are distributed in the solvendum
Substitutional crystalline solid solutions: A substitution crystalline solid dispersion is a type of solid solutions which is having a crystalline structure, in that the solute molecules substitute for solvent molecules in the crystal lattice. Substitution is only possible when the size of the solute molecules differs by less than 15% or so from that of the solvent molecules (Fig. 6) Substitutional solid solution.
Interstitial crystalline solid solutions: In interstitial solid solutions, dissolved molecules occupy the interstitial spaces between the solvent molecules in the crystal lattice. As in the case of substitutional crystalline solid solutions, the relative molecular size is a crucial criterion for classifying the solid solution type.
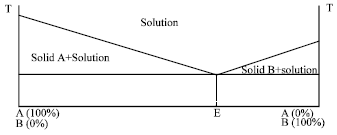 | |
| Fig. 4: | Hypothetical phase diagram of a continuous solid solution (Giri et al., 2010) |
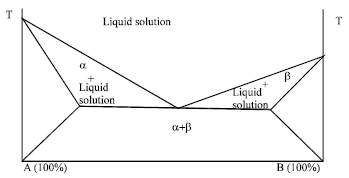 | |
| Fig. 5: | Hypothetical phase diagram of a discontinuous solid solution (Goldberg et al., 1965) |
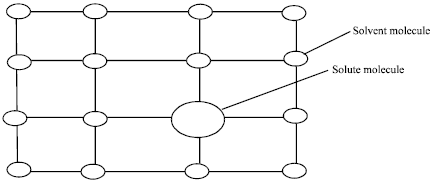 | |
| Fig. 6: | Substitutional solid solution (Maski et al., 2009) |
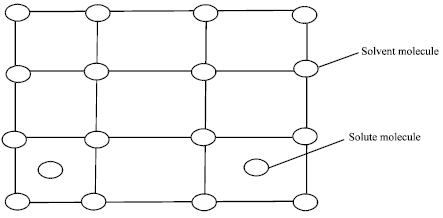 | |
| Fig. 7: | Interstitial solid solution |
 | |
| Fig. 8: | Amorphous solid solution (Suryawanshi et al., 2010) |
In the case of interstitial crystalline solid solutions, the solute molecules should have a molecular diameter that is no greater than 0.59 of the solvent molecule's molecular diameter Furthermore, the volume of the solute molecules should be less than 20% of the solvent (Fig. 7) Interstitial solid solution.
Amorphous solid solution: It is demonstrated, that drug with propensity to super cooling has more tendency to solidify as an amorphous form in presence of carrier (Nikhil, 2010). This is quite similar to simple eutectic mixtures but only difference is that drug is precipitated out in an amorphous form. Ex. Precipitation of sulfathiazole in crystalline urea (Fig. 8), amorphous solid solution (Table 4) and classification of Solid Dispersions according to Molecular arrangement (Gavali et al., 2011).
| Table 4: | Classification of solid dispersions according to molecular arrangement (Sonpal et al., 2011) |
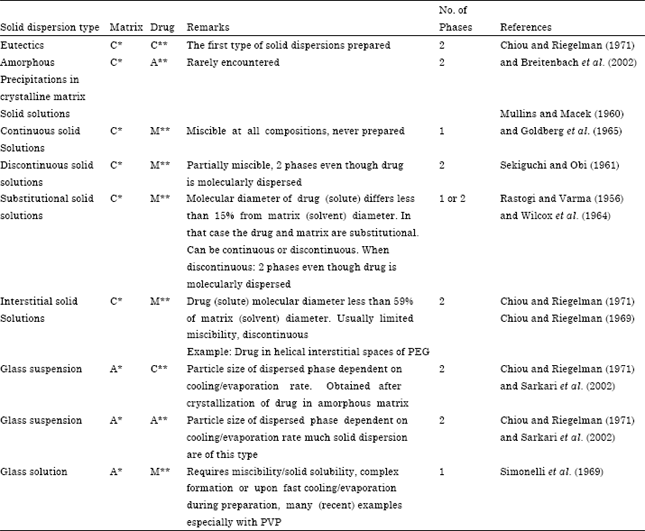 | |
| A*: Matrix in the amorphous state, C*: Matrix in the crystalline state, A**: Drug dispersed as amorphous clusters in the matrix, C**: Drug dispersed as crystalline particles in the matrix, M**: Drug molecularly dispersed throughout the matrix | |
Glass solutions: Solute dissolves in glass carrier to form a homogeneous glassy system is known as glass solutions (Swarbrick, 2006; Kim et al., 2010). Glass suspensions are mixture in which precipitated particles are suspended in glass solvent. Different characteristics of glassy state are brittleness, transparency below the glass transition temperature. Lattice energy (barrier to rapid dissolution) is much lower in glass solution and suspension. Ex-Carriers for glass solution and suspension-citric acid, sugars (dextrose, sucrose and galactose), PVP, PEG and urea (British Pharmacopoeia, 2007; Van Drooge et al., 2004) (Table 4) Different carriers used for the preparation of solid dispersion (Naveen et al., 2010) (Fig. 9) Schematic picture of the variation of enthalpy (or volume) with temperature. TG-glass transition temp, T f-M.P. of material.
METHODS OF PREPARATION OF SOLID DISPERSIONS
Hot melt method: A process of transferring a powder blend of drug and carrier by a rotating screw, through the heated barrel of an extruder and pressing the melt through a die into a product of uniform shape is known as Hot-Melt Extrusion (HME) or fusion method (McGinity and Zhang, 2003).
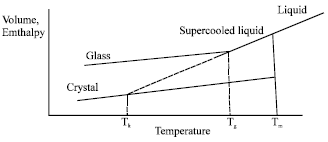 | |
| Fig. 9: | Schematic picture of the variation of enthalpy (or volume) with temperature (Shujun et al., 2006) Tag: Glass transition temp, To: M.P. of material |
HME was first introduced in the plastics industry in the mid-nineteenth century to apply polymeric insulation coatings to wires (Crowley et al., 2007). First applications of HME were realized as a manufacturing tool in the pharmaceutical industry (Chaudhuri, 2007).
Solvent evaporation method: The process which involve solubilization of drug and carrier in a volatile solvent which is later evaporated is termed as Solvent Evaporation Method (SEM). (Hasegawa et al., 1985; Lloyd et al., 1999; Lima et al., 2008). In this method, the thermal decomposition of drugs or carriers can be prevented, since organic solvent evaporation occurs at low temperature (Won et al., 2005; Gupta et al., 2008; Singh et al., 2011). Solvent evaporation method is popularly used for preparation of microsphere because of its simplicity, fast processing and reproducibility with minimum controllable process variables that can be easily implemented at the industrial level. Many studies have been done on solid dispersions of Meloxicam (Chokshi and Hossein, 2004; Leila et al., 2011), Naproxen and (Mullins and Macek, 1960), by solvent evaporation techniques.
Fusion method: A method in which a molten mixture of drug and carrier are cooled to solidification, is called as fusion method it is also called as solvent method in which precipitation of drug and carrier from a common solvent occur. Paracetamol solid dispersion with PEG 8000 was prepared by melt fusion method (Khan et al., 2011).
Melting solvent method: This involves dissolution of drug in a minimum amount of an organic solvent, which is then added to the molten carrier (Chiou and Riegelman, 1969; Gupta and Moorthy, 2007). Melting solvent method (melt evaporation) method is used to prepare spironolactone-polyethylene glycol 6000 solid dispersion without removing the solvent. They mention that 5-10% (w/w) of liquid compound could be incorporated into polyethylene glycol 6000 without significant loss of its solid property (Table 5). Some resent patent on solubility enhancement using solid dispersion technique (Schroeder, 2009; Ajazuddin and Saraf, 2011) has been shown in Table 5.
COMPLEXATION
Cyclodextrins (CD) is a group of cyclic oligosaccharides, known for their ability to form inclusion complexes with a variety of organic molecules (Saenger et al., 1984; Khan et al., 2001) Complexation by Cyclodextrins, especially the most commonly available β-Cyclodextrins, is widely used to increase the solubility of drug molecules which have limited solubilities in water (Abou-Auda et al., 2006).
| Table 5: | Some resent patent on solubility enhancement using solid dispersion technique (Schroeder, 2009) |
 | |
| Table 6: | List of complexing agents |
 | |
Cyclodextrins Can also be used to prevent drug-drug interaction, it Convert liquid drug in to microcrystalline powders, decreases volatility, modify gastrointestinal or ocular irritation and mask of objectionable taste or odor of drug. Cyclodextrins of pharmaceutical relevance contain 6, 7 or 8 dextrose molecules (α, β, γ-Cyclodextrins) bound in a 1, 4-configuration to form rings of various diameters. The ring has a hydrophilic exterior and lipophilic core in which appropriately sized organic molecules can form noncovalent inclusion complexes resulting in increased aqueous solubility and chemical stability. Complexation is occurring between two or more molecules to form a nonbonded entity with a well defined stoichiometry. Complexation relies weak forces such as London forces, hydrogen bonding and hydrophobic interactions. The Inclusion complexes can induce modification of the physicochemical properties of the guest molecules, particularly in terms of water solubility and solution stability (Lyng et al., 2004). Complex Formation by Cyclodextrins shown in Fig. 10 (Khan et al., 2001). Different method are used to prepare inclusion complexes of a variety of drugs in order to improve their solubility and dissolution rate. E.g., Co-precipitation, kneading and solid dispersion methods (Shujun et al., 2006). There are many types of complexing agents and a partial list can be found in Table 6.
SOLUBILISATION BY SURFACTANTS
Surfactants are known to play a vital role in pharmacy because it have an ability to increase the solubility of poorly soluble drug in water (Gharaei-Fathabad, 2011; Moghaddam and Moghaddam, 2011).
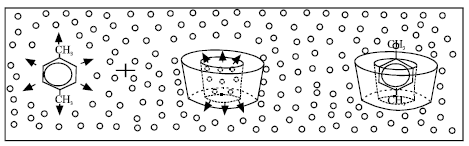 | |
| Fig. 10: | Complex Formation by Cyclodextrins (Kawashima et al., 1975) |
One of important property of surfactants is the formation of colloidal-sized clusters in solutions, called as micelles which is having a particular significance in pharmacy. Surfactant having the characteristic property of reducing the interfacial and surface tension using the same mechanism as chemical surfactant. Surfactants are the molecules with distinct no Polar Regions (Emara et al., 2002). Most surfactants consist of a hydrocarbon segment connected to a polar group. The polar group can be cationic, anionic, nonionic or zwitterionic. When small polar molecules are added they can accumulate in the hydrophobic core of the micelles. This technique of solubilization is very important in biological and industrial processes (Gavali et al., 2011). This work was investigated to develop the carvedilol tablets, allowing fast, reproducible and complete drug dissolution, by using surfactant.
Microemulsions: The concept of microemulsion was first introduced by Hoar and Schulman (1943). A monodispersion spherical droplets consisting of oil, surfactant, co-surfactant and aqueous phase, which is optically isotropic and thermodynamically stable with a droplet diameter within the range of 10-100 nm is defined as ‘microemulsion (Tenjarla, 1999; Yazdani and Hadianfard, 2012). Microemulsions could enhance the potential solubilization of hydrophobic drugs (Yin et al., 2009; Alexander et al., 2011a). Amongst the various drug delivery systems, the microemulsion system is considered as an ideal alternative for the oral delivery of lipophilic drug.
Self micro emulsifying drug delivery systems: For the improving solubility, dissolution and oral absorption of hydrophobic drugs ‘self- micro emulsifying drug delivery systems’ (SMEDDS) have been preferred (Breitenbach et al., 2002; Cui et al., 2005). SMEDDS is a isotropic mixtures of an oil, surfactant, co surfactant or (solubilizer) and drug. The basic principle of this system is its ability to form fine oil-in-water (o/w) microemulsions under gentle agitation following dilution by aqueous phases.
Self emulsifying drug delivery systems (SEDDS): An isotropic mixture of oils, surfactants, along with co-solvents/surfactants that have a unique ability of forming fine oil-in-water (o/w) micro emulsions upon moderate mixing of these ingredients in aqueous media, such as GI (Gastro Intestinal) fluids is termed as Self Emulsifying Drug Delivery Systems (SEDDS) (Agrawal et al., 2012). It is the most useful technology to improve the rate and extent of this poorly water soluble drug. SEDDS is a mixture of oil, surfactant and if necessary a solubiliser. Self emulsification is initiated under gentle agitation following contact with aqueous phase and forms a thermodynamically stable o/w microemulsion with particle diameter of 100 nm or less. They are reputed to improve the oral bioavailability of poorly water soluble drug (Obitte et al., 2008; Ajazuddin and Saraf, 2010b).
CHEMICAL MODIFICATIONS (Rytting et al., 2005)
Salt formation: For enhancement solubility and dissolution rates of acidic and basic drugs salt formation is the most common and effective method (Serajuddin, 2007). Salts of acidic and basic drugs have, in general, higher solubility than their corresponding acid or base forms. Salt formation to enhance the aqueous solubility is the most preferred approach for the development of liquid formulations for parenteral administration (Sweetana and Akers, 1996; Lakade and Bhalekar, 2010).
Co-crystallization: The crystalline material that consists of two or more molecular and electrical neutral species held together by non-covalent forces is termed as co-crystallization’ (Masuda et al., 2012). The non-ionizable drugs can be form due to the co crystal, which cannot undergo in salt formation (Childs et al., 2007). By the addition, for ionizable drugs, the number of suitable co crystal formers can exceed the number of suitable salt formers. For example, the ionizable drug piroxicam has more than 50 reported co crystal formers (Tran et al., 2010).
Co-solvent: Non-aqueous co-solvent systems have been evaluated for their potential use in the freeze-drying of pharmaceutical products. Co-solvents have been reported to affect the rate of the organic phase partitioning into the external aqueous phase and thus, influence the physicochemical properties and release kinetics of PLGA microspheres (Rudra et al., 2011; Singh et al., 2011).
Hydrotropic: For drug aqueous solubility ‘hydrotropic’ solubilization is an important technique (Shibata et al., 2009). Since 1916, New berg, was first suggested the term hydrotropic which is used to designate anionic organic salts which, at high concentrations, considerably increase the aqueous solubility of poorly soluble solutes. Hydrotropes dissolved in water which can produce high degree solubility enhancement of hydrophobic drugs (Trana et al., 2011). For enhancement of aqueous solubility of hydrophobic drugs ‘hydrotropic agents’ have been found to be more effective and hence can play important role for improving the oral bioavailability (Maheshwari and Jagwani, 2011; Alexander et al., 2011b) (Table 7). Hydrotropic is a molecular phenomenon where by adding a second solute (the hydrotropic) results in an increase in the aqueous solubility of poorly soluble solutes (Nidhi et al., 2011) (Table 8) provide the example of some drug which Enhance the solubility by using various technique.
| Table 7: | Hydrotropic is a molecular phenomenon where by adding a second solute results in an increase in the aqueous solubility of poorly soluble solutes (Bobe et al., 2011) |
 | |
| Table 8: | Example of some drug which Enhance the solubility by this technique |
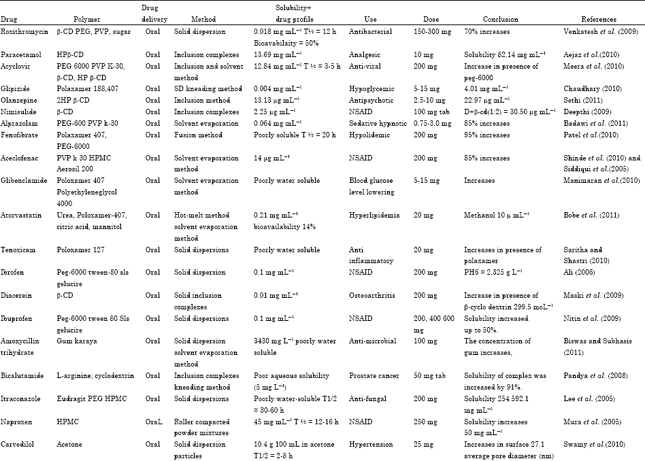 | |
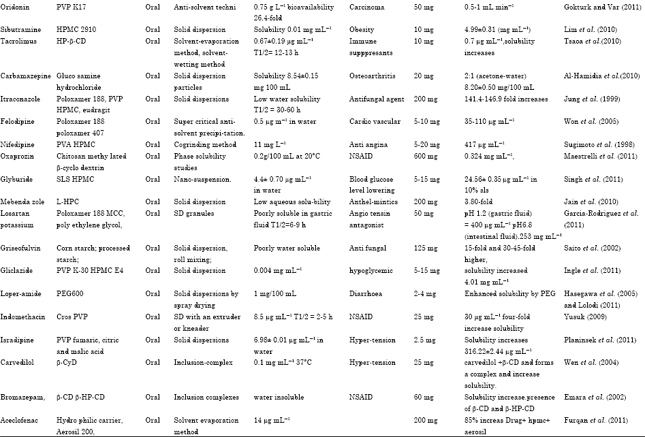 | |
CONCLUSION
The stability of the drug, its solubility and availability at the site of action, is very important particularly when the formulation is intended for oral administration. Solubility and dissolution can be subsequently affecting the in vivo absorption of drug. So, it is very important to improve the aqueous solubility drugs. By reviewing this article we conclude that, solubility is a most important parameter for the oral bioavailability of hydrophobic. Solubility is also the basic requirement for the formulation and development of different dosage form of different drugs. Solubility can be enhanced by many techniques and number of folds increase in solubility is reported too. Because due to the solubility and stability problem of many drugs the bioavailability of them gets affected and hence solubility enhancement becomes necessary. It is now possible that to increase the solubility of hydrophobic drugs with the help of various techniques as mentioned above.
ACKNOWLEDGEMENT
The authors would like to acknowledge the assistance provided by the Library of “Rugnta College of Pharmaceutical Sciences and Research, Kohka-kurud Road, Bhiali, C.G. (India) for collection of literature.
REFERENCES
- Ajazuddin and S. Saraf, 2010. Evaluation of physicochemical and phytochemical properties of Safoof-E-Sana, a Unani polyherbal formulation. Pharmacogn. Res., 2: 318-322.
CrossRefDirect Link - Amit, A., R. Chaurasia, J. Khan, Swarna, S. Sahu and S. Patel, 2011. Spectrophotometric method of standard curve preparation and calculation for metronidazole. Int. J. Pharm. Professional's Res., 2: 206-209.
Direct Link - Alexander, A., Ajazuddin, D.K. Tripathi, T. Verma, Swarna, J. Maurya and S. Patel, 2011. Mechanism responsible for mucoadhesion of mucoadhesive drug delivery system: A review. Int. J. Applied Biol. Pharm. Technol., 2: 434-445.
Direct Link - Alexander, Amit Sharad Sharma, Ajazuddin, Khan Mohammed Junaid and Swarna, 2011. Theories and factors affecting mucoadhesive drug delivery systems: A review. Int. J. Ayurveda Pharm., 2: 1155-1161.
Direct Link - Alexander, A., Ajazuddin, D.K. Tripathi, V. Tekeshwar, P. Sandip, D. Harsh and Swarna, 2011. Role of excipients to enhance the disintegration property of different formulations: An Overview. Res. J. Pharmacy Technol., 4: 1519-1525.
Direct Link - Al-Haj, N. and A. Rasedee, 2009. Solid lipid nanoparticles preparation and characterization. Int. J. Pharmacol., 5: 90-93.
CrossRefDirect Link - Badawi, A.A., M.A. El-Nabarawi, D.A. El-Setouhy and S.A. Alsammit, 2011. Characterization and stability testing of itraconazole solid dispersions containing crystallization inhibitors. Am. J. Drug Discovery Dev., 1: 144-159.
CrossRef - Amarji, B., Ajazuddin, D. Raghuwanshi, S.P. Vyas and P. Kanaujia, 2007. Lipid Nano Spheres (LNSs) for enhanced oral bioavailability of amphotericin B: Development and characterization. J. Biomed. Nanotechnol., 3: 264-269.
Direct Link - Amidon, G.L., H. Lennernaes, V.P. Shah and J.R. Crison, 1995. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res., 12: 413-420.
CrossRef - Blagden, N., M. de Matas, P.T. Gavan and P. York, 2007. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Delivery Rev., 59: 617-630.
CrossRefDirect Link - Chattopadhyay, A.B., S. Thomas and R. Chatterjee, 2011. Analysis of steady state stability of a Csi fed synchronous motor drive system with damper windings included. Applied Sci. Res., 6: 992-1005.
CrossRef - Chaudhuri, A.A., 2007. Construction of a rational delta function using the reverse cantor set and its application to quantum mechanics via pseudo-spectral methods. Trends Applied Sci. Res., 2: 1-14.
CrossRefDirect Link - Chiou, W.L. and S. Riegelman, 1971. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci., 60: 1281-1302.
CrossRef - Chiou, W.L. and S. Riegelman, 1969. Preparation and dissolution characteristics of several fast-release solid dispersions of griseofulvin. J. Pharm. Sci., 58: 1505-1510.
CrossRefPubMedDirect Link - Chokshi, R. and H. Zia, 2004. Hot-melt extrusion technique: A review. Iran J. Pharm. Res., 3: 3-16.
Direct Link - Leaner, C. and J. Dressman, 2000. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm., 50: 47-60.
CrossRef - Davis, M., C.J. Simmons, B. Dordoni and R. Williams, 1974. Urinary D-glucaric acid excretion and plasma antipyrine kinetics during enzyme induction. Br. J. Clin. Pharmacol., 1: 253-257.
Direct Link - Lim, H.T., P. Balakrishnan, D.H. Oh, K.H. Joe and Y.R. Kim et al., 2010. Development of novel sibutramine base-loaded solid dispersion with gelatin and HPMC: Physicochemical characterization and pharmacokinetics in beagle dogs. Int. J. Pharma., 397: 225-230.
Direct Link - Floyd, A.G., 1999. Top ten considerations in the development of parenteral emulsions. Pharm. Sci. Technol. Today, 2: 134-143.
CrossRef - Goldberg, A.H., M. Gibaldi and J.L. Kanig, 1965. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures I. Theoretical considerations and discussion of the literature. J. Pharm. Sci., 54: 1145-1148.
CrossRef - Goldberg, A.H., M. Gibaldi and J.L. Kanig, 1966. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures II: Experimental evaluation of a eutectic mixture: Urea-acetaminophen system. J. Pharm. Sci., 55: 482-487.
CrossRef - Grau, M.J., O. Kayser and R.H. Muller, 2000. Nanosuspensions of poorly soluble drugs- reproducibility of small scale production. Int. J. Pharm., 196: 155-159.
Direct Link - Gupta, U., H.B. Agashe, A. Asthana and N.K. Jain, 2006. Dendrimers: Novel polymeric nanoarchitectures for solubility enhancement. Biomacromolecules, 7: 649-658.
CrossRef - Hasegawa, S., T. Hamaura, N. Furuyama, A. Kusai, E. Yonemochi and K. Terada, 2005. Effects of water content in physical mixture and heating temperature on crystallinity of troglitazone-PVP K30 solid dispersions prepared by closed melting method. Int. J. Pharm., 302: 103-112.
CrossRefPubMedDirect Link - Heimbach, T., D. Fleisher and A. Kaddoumi, 2007. Overcoming poor aqueous solubility of drugs for oral delivery. Prodrugs, 5: 157-215.
CrossRef - Uchiyama, H., Y. Tozuka, F. Asamoto and H. Takeuchi, 2011. α-Glucosyl hesperidin induced an improvement in the bioavailability of pranlukast hemihydrate using high-pressure homogenization. Int. J. Pharma., 410: 114-117.
PubMed - Abou-Auda, H.S., S.A. Bawazir, Y.A. Asiri, O.A. Gubara and B.M. AI-Hadiya, 2006. Studies on solubility, bioavability and hypoglycemic activity of gliclazide β-cyclodextrins complexes. Int. J. Pharmacol., 2: 656-663.
Direct Link - Jung, J.Y., S.D. Yoo, S.H. Lee, K.H. Kim, D.S. Yoon and Kyu-Hyun Lee. 1999. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. Int. J. Pharm., 187: 209-218.
Direct Link - Kim, J.Y., S. Kim, R. Pinal and K. Park, 2011. Hydrotropic polymer micelles as versatile vehicles for delivery of poorly water-soluble drugs. J. Control. Release., 152: 13-20.
PubMed - Kim, J.Y., S. Kim, M. Papp, K. park and R. Pinal, 2010. Hydrotropic solubilization of poorly water-soluble drugs. J. Pharm. Sci., 99: 3953-3965.
Direct Link - Jounela, A., P. Pentikainen and A. Sothmann, 1975. Effect of particle size on the bioavailability of digoxin. Eur. J. Clin. Pharmacol., 8: 365-370.
PubMed - Garcia-Rodriguez, J.J., P.M. de la Torre-Iglesias, M.C. Vegas-Sanchez, S. Torrado-Duran, F. Bolas-Fernandez and S. Torrado-Santiago, 2011. Changed crystallinity of mebendazole solid dispersion: Improved anthelmintic activity. Int J. Pharma., 403: 23-28.
PubMed - Tsaoa, J.Y., H.H. Tsai, C.P. Wu, P.Y. Lin and S.Y. Su et al., 2010. Release of paeonol β-CD complex from thermo-sensitive poly (N-isopropylacrylamide) hydrogels. Int. J. Pharm., 402: 123-128.
PubMed - Kawashima, Y., M. Saito and H. Takenaka, 1975. Improvement of solubility and dissolution rate of poorly water-soluble salicylic acid by a spray-drying technique. J. Pharm. Pharmacol., 27: 1-5.
CrossRef - Khan, G.M., F. Wazir and J.B. Zhu, 2001. Ibuprofen-β-cyclodextrin inclusion complexes: Evaluation of different complexation methods. J. Medical Sci., 1: 193-199.
CrossRefDirect Link - Lindenberg, M., S. Kopp and J.B. Dressman, 2004. Classification of orally administered drugs on the world health organization model list of essential medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm., 58: 265-278.
CrossRefPubMedDirect Link - Liversidge, G.G. and K.C. Cundy, 1995. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharmaceut., 125: 91-97.
CrossRefDirect Link - Lokhande, P.D., K.R. Gawai, K.M. Kodam, B.Y. Waghmare, A.R. Chabukswar and S.C. Jagdale, 2006. Water soluble amide derivatives of polyene antibiotic and their antifungal activity. Trends Applied Sci. Res., 1: 529-533.
CrossRefDirect Link - Lloyd, G.R., D.Q.M. Craig and A. Smith, 1999. A calorimetric investigation into the interaction between paracetamol and polyethlene glycol 4000 in physical mixes and solid dispersions. Eur. J. Pharm. Biopharm., 48: 59-65.
CrossRef - Maestrelli, F., M. Cirri, N. Mennini, N. Zerrouk and P. Mura, 2011. Improvement of oxaprozin solubility and permeability by the combined use of cyclodextrin, chitosan and bile components. Eur. J. Pharm. Biopharm., 78: 385-393.
CrossRef - Maheshwari, R.K. and Y. Jagwani, 2011. Mixed hydrotropy: Novel science of solubility enhancement. Indian J. Pharm. Sci., 73: 179-183.
CrossRefPubMedDirect Link - Al Omari, M.M., N.H. Daraghmeh, M.I. El-Barghouthi, M.B. Zughul, B.Z. Chowdhry, S. Leharne and A.A. Badwan, 2009. Novel inclusion complex of ibuprofen tromethamine with cyclodextrins: Physico-chemical characterization. J. Pharm. Biomed. Anal., 50: 449-458.
CrossRef - Malpani, A.S., P. Waghere and N.V. Belorkar, 2009. Aqueous solubility: Measurement and prediction tools. Latest Rev., 7: 1-10.
Direct Link - Manimaran, V., N. Damodharan, M. Mothilal, K. Rajkumar and R.M. Chalackal, 2010. Enhancement of dissolution rate of glibenclamide by solid dispersion technology. Int. J. Curr. Pharm. Res., 2: 14-17.
Direct Link - Sugimoto, M., T. Okagaki, S. Narisawa, Y. Koida and K. Nakajima, 1998. Improvement of dissolution characteristics and bioavailability of poorly water-soluble drugs by novel cogrinding method using water-soluble polymer. Int. J. Pharm., 160: 11-19.
CrossRefDirect Link - Saito, M., T. Ugajin, Y. Nozawa , Y. Sadzuka, A. Miyagishima and T. Sonobe, 2002. Preparation and dissolution characteristics of griseofulvin solid dispersions with saccharides. Int. J. Pharm., 249: 71-79.
CrossRef - Maski, N., Arulkumaran, K. Girhepunje, P. Ghode, S. Randive and Ranju Pal, 2009. Studies on the preparation, characterization and solubility of β-cyclodextrin-diacerein inclusion complexes. Int. J. Pharmacy Pharmaceut. Sci., 1: 121-135.
Direct Link - Deepthi, M., 2009. A study on suitability of nimesulide-betacyclodextrin complex in oral and topical dosage forms. Int. J. Pharm. Pharma. Sci., 1: 193-198.
Direct Link - Mehnert, W. and K. Mader, 2001. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev., 47: 165-196.
CrossRef - Dabbagh, M.A. and B. Taghipour, 2007. Investigation of solid dispersion technique in improvement of physicochemical characteristics of ibuprofen powder. Iran. J. Pharm. Sci., 3: 69-76.
Direct Link - Mosharraf, M. and C. Nystrom, 1995. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int. J. Pharm., 122: 35-47.
CrossRef - Muller, R.H., C. Jacobs and O. Kayser, 2001. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Delivery Rev., 47: 3-19.
CrossRef - Mullins, J.D. and T.J. Macek, 1960. Some pharmaceutical properties of novobiocin. J. Am. Pharm. Assoc., 49: 245-248.
CrossRef - Wu, W. and G.H. Nancollas, 1998. A new understanding of the relationship between solubility and particle size. J. Solution Chem., 27: 521-531.
CrossRef - Noorizadeh, H. and A. Farmany, 2011. Investigation of capacity behaviors by linear and nonlinear models chemometrics. Trends Applied Sci. Res., 6: 1324-1334.
CrossRef - Leila, N., H. Sakina, B. Abdelaziz, M. Fatiha and L.L.D. Fateh, 2011. Theoretical study of the inclusion processes of the phenylurea herbicide metobromuron in β-cyclodextrin. J. Biol. Sci., 11: 1-9.
CrossRefDirect Link - Nourani, V., A.A. Moghaddam, A.O. Nadiri and V.P. Singh, 2008. Forecasting spatiotemporal water levels of tabriz aquifer. Trends Applied Sci. Res., 3: 319-329.
CrossRefDirect Link - Obitte, N.C., H. Ezeiruaku and V.I. Onyishi, 2008. Preliminary studies on two vegetable oil based self emulsifying drug delivery system (SEDDS) for the delivery of metronidazole, a poorly water soluble drug. J. Applied Sci., 8: 1950-1955.
CrossRefDirect Link - Planinsek, O., B. Kovacic and F. Vrecer, 2011. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int. J. Pharm., 406: 41-48.
CrossRef - Patidar, K., S. Manish, S.K. Dinesh and J.K. Surendra, 2010. Solid dispersion: Approaches, technology involved, unmet need and challenges. Drug Invention Today, 2: 349-357.
Direct Link - Porter, C.H.J. and W.N. Charman, 2001. Intestinal lymphatic drug transport: An update. Adv. Drug Delivery Rev., 50: 61-80.
CrossRef - Gupta, S.P.B.N. and N.S.H.N. Moorthy, 2007. Synthesis and physicochemical characterization of mutual prodrug of indomethacin. Trends Applied Sci. Res., 2: 165-169.
CrossRefDirect Link - Rainbow, B.E., 2004. Nanosuspensions in drug delivery. Nat. Rev. Drug. Discovery , 3: 785-796.
CrossRef - Rastogi, R.P. and K.T.R. Varma, 1956. Solid-liquid equilibria in solutions of non-electrolytes. J. Chem. Soc., 2: 2097-2101.
CrossRefDirect Link - Rodier, E., H. Lochard, M. Sauceau, J.J. Letourneau, B. Freiss and J. Fages, 2005. A three step supercritical process to improve the dissolution rate of Eflucimibe. Eur. J. Pharm. Sci., 26: 184-193.
CrossRef - Rudra, A., K. Santra and B. Mukherjee, 2011. Poly [D, L-lactide-co-glycolide] microspheres as a delivery system of protein ovalbumin used as a model protein drug. Trends Applied Sci. Res., 6: 47-56.
CrossRefDirect Link - Singh, S.K., K.K. Srinivasan, K. Gowthamarajan, D.S. Singare, D. Prakash and N.B. Gaikwad, 2011. Investigation of preparation parameters of nanosuspension by top- down media milling to improve the dissolution of poorly water-soluble glyburide. Eur. J. Pharm. Biopharm., 78: 441-446.
PubMed - Gavali, S.M., S.S. Pacharane, S.V. Sankpal, K.R. Jadhav and V.J. Kadam, 2011. liquisolid compact: A new technique for enhancement of drug dissolution. IJRPC, 1: 705-713.
Direct Link - Lakade, S.H. and M.R. Bhalekar, 2010. Different types of method for modified dosage form for enhancement of dissolution rate through solid dispersion. Int. J. Pharma. Stud. Res., 1: 54-63.
Direct Link - Sangshetti, J.N., P.R. Mahaparale, S. Paramane and D.B. Shinde, 2008. spectrophotometric estimation of donepezil hydrochloride in bulk and tablet formulation. Trends Applied Sci. Res., 3: 109-112.
CrossRefDirect Link - Keiji, S. and O. Noboru, 1961. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharm. Bull., 9: 866-872.
Direct Link - Serajuddin, A.T.M., 2007. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev., 59: 603-616.
CrossRefDirect Link - Sharma, D., M. Soni, S. Kumar and G.D. Gupta, 2009. Solubility enhancement-eminent role in poorly soluble drugs. Res. J. Pharm. Tech., 2: 220-224.
Direct Link - Shinde, S.S., S.S. Patil, F.I. Mevekari and A.S. Satpute, 2010. An approach for solubility enhancement: Solid dispersion. Int. J. Adv. Pharm. Sci., 1: 299-308.
Direct Link - Moghaddam, S.S. and M.S. Moghaddam, 2011. A comprehensive survey on antenna array signal processing. Trends Applied Sci. Res., 6: 507-536.
CrossRefDirect Link - Shujun, W., Y. Jinglin and G. Wenyuan, 2006. Use of X-ray diffractometry for identification of different Fritillaria traditional Chinese medicine. Trends Applied Sci. Res., 1: 334-340.
CrossRefDirect Link - Lee, S., K. Nam, M.S. Kim, S.W. Jun, J.S. Park, J.S. Woo and S.J. Hwang, 2005. Preparation and characterization of solid dispersions of itraconazole by using aerosol solvent extraction system for improvement in drug solubility and bioavailability. Arch. Pharm. Res., 28: 866-874.
PubMed - Gokturk, S. and U. Var, 2011. Effect of ethanol on partition and binding equilibrium of phenothiazine in anionic and nonionic micellar solutions. Curr. Res. Chem., 3: 49-61.
CrossRefDirect Link - Strickley, R.G., 2004. Solubilizing excipients in oral and injectable formulations. Pharm. Res., 21: 201-230.
CrossRefDirect Link - Swamy, P.V., H. Shilpa, S.B. Shirsand, S.N. Gada and M.B. Kinagi, 2010. Role of cogrinding in enhancing the In vitro dissolution characteristics of carvedilol. Int. J. Pharm. Sci. Res., 1: 232-237.
Direct Link - Sweetana, S. and M.J. Akers, 1996. Solubility principles and practices for parenteral dosage form development. PDA J. Pharm. Sci. Technol., 50: 330-342.
Direct Link - Masuda, T., Y. Yoshihashi, E. Yonemochi, K. Fujii, H. Uekusa and K. Terada, 2012. Cocrystallization and amorphization induced by drug-excipient interaction improves the physical properties of acyclovir. Int. J. Pharma., 422: 160-169.
CrossRef - Tapas, A.R., P.S. Kawtikwar and D.M. Sakarkar, 2011. Modification of felodipine properties using spherically agglomerated solid dispersions. Am. J. Drug Discovery Dev., 1: 160-173.
CrossRefDirect Link - Patel, T., L.D. Patel, T. Patel, S. Makwana and T. Patel, 2010. Enhancement of dissolution of Fenofibrate by Solid dispersion Technique. Int. J. Res. Pharm. Sci., 1: 127-132.
Direct Link - Tenjarla, S., 1999. Microemulsions: An overview and pharmaceutical applications. Crit. Rev. Ther. Drug Carrier Syst., 16: 461-521.
PubMed - Tran, T.T., P.H. Tran, H.G. Choi, H.K. Han and B.J. Lee, 2010. The roles of acidifiers in solid dispersions and physical mixtures. Int. J. Pharm., 384: 60-66.
CrossRefPubMedDirect Link - Tian, L., H. He and X. Tang, 2007. Stability and degradation kinetics of etoposide-loaded parenteral lipid emulsion. J. Pharm. Sci., 96: 1719-1728.
PubMed - Vahedi, H., 2012. Double band adaptive hysteresis current control employed in active power filter. Trends Applied Sci. Res., 7: 151-159.
CrossRefDirect Link - Vyas, A., S. Saraf and S. Saraf, 2008. Cyclodextrin based novel drug delivery systems. J. Inclusion Phenomena Macrocyclic Chem., 62: 23-42.
CrossRefDirect Link - Westesen, K., H. Bunjes and M.H.J. Koch, 1997. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. Control Release, 48: 223-236.
CrossRefDirect Link - Won, D.H., M.S. Kim, S. Lee, J.S. Park and S.J. Hwang, 2005. Improved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation process. Int. J. Pharma., 301: 199-208.
CrossRefPubMedDirect Link - Wen, X., F. Tan, Z. Jing and Z. Liu, 2004. Preparation and study the 1:2 inclusion complex of carvedilol with β-cyclodextrins. J. Pharm. Biomed. Anal., 34: 517-523.
Direct Link - Yazdani, A. and M.J. Hadianfard, 2012. A new model in glass forming range of binary alloys. Trends Applied Sci. Res., 7: 175-180.
CrossRefDirect Link - Yin, Y.M., F.D. Cui, C.F. Mu, M.K. Choi and J.S. Kim et al., 2009. Docetaxel microemulsion for enhanced oral bioavailability: Preparation and in vitro and in vivo evaluation. J. Control Release, 140: 86-94.
CrossRefPubMedDirect Link - Shibata, Y., M. Fujii, Y. Sugamura, R. Yoshikawa and S. Fujimoto et al., 2009. The preparation of a solid dispersion powder of indomethacin with crospovidone using a twin-screw extruder or kneader. Int. J. Pharma., 365: 53-60.
CrossRefPubMedDirect Link - Zhixun, L., F. Yan and Z. Guozhong, 2006. The applying of THz and Raman techniques in non-destructive examination for nitrobenzoic acid. Trends Applied Sci. Res., 1: 176-183.
CrossRefDirect Link - Seedher, N. and P. Sharma, 2007. Solubility and stability enhancement of poorly-soluble drugs clarithromycin and prednisolone by combination with other drugs. Int. J. Biol. Chem., 1: 229-236.
CrossRefDirect Link - Agrawal, S., T. Giri, D.K. Tripathi, Ajazuddin and A. Alexander, 2012. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant based Self Micro Emulsifying Drug Delivery System (SMEDDS): A novel approach. Am. J. Drug Discovery Develop., (In Press).
- Nidhi, K., S. Indrajeet, M. Khushboo, K. Gauri and D.J. Sen, 2011. Hydrotropy: A promising tool for solubility enhancement: A review. Int. J. Drug Dev. Res., 3: 26-33.
Direct Link - Breitenbach, J., 2002. Melt extrusion: From process to drug delivery technology. Eur. J. Pharm. Biopharm., 54: 107-117.
CrossRef - Ugazio, E., R. Cavalli and M.R. Gasco, 2002. Incorporation of cyclosporin A in Solid Lipid Nanoparticles (SLN). Int. J. Pharm., 241: 341-344.
CrossRef