
Febby J. Polnaya
Faculty of Agriculture, Pattimura University, Jl. Ir. M. Putuhena, Kampus Poka Ambon, 97233 Maluku, Indonesia
Djagal W. Marseno
Faculty of Agricultural Technology, Gadjah Mada University, Jl. Flora No. 1 Bulaksumur, 55281 Yogyakarta, Indonesia
Muhammad N. Cahyanto
Faculty of Agricultural Technology, Gadjah Mada University, Jl. Flora No. 1 Bulaksumur, 55281 Yogyakarta, Indonesia
Pakistan Journal of Nutrition
Year: 2018 | Volume: 17 | Issue: 4 | Page No.: 199-206







ABSTRACT
Background and Objective: Native sago starch (NSS) was phosphorylated white sodium tripolyphosphate (STPP) or phosphorous oxychloride (POCl3) to increase the properties of starch. The phosphorylated sago starch play a significant role in forming new structure that affect the physical properties and in vitro digestibility of resistant starch (RS). The objective of this study was to investigate the effects of the reaction of NSS with STPP or POCl3 at pH 8, 9, 10 and 11 on the physical properties and in vitro digestibility of RS. Materials and Methods: The RS from NSS was prepared by phosphorylating with 5% STPP or 4% POCl3 at pH 8-11 for 1 h. The functional groups, relative degree of cross-linking, degree of crystallinity of selected RS samples and RS contents, were also investigated. Results: The FT-IR spectra showed that after being hydrolyzed, RS contains α,γ-dextrin enriched with phosphate groups. The di-starch phosphate had relative degrees of cross-linking ranging from 22.83-85.33%, while that mono-starch phosphate had not able to estimate. The XRD pattern indicated that the crystalline structure of RS was destroyed. The mono- and di-starch phosphate had degrees of crystallinity ranging from 6.53-6.64%. Compared with NSS, the RS contents of mono- and di-starch phosphate were higher by 20 and 40%, respectively. The RS content of phosphorylated starch was affected by the substitution of phosphate groups. Conclusion: The RS from mono- and di-starch phosphate were affected by the presence of the phosphorus group in the structure and the RS content could be improved greatly with phosphorylation. The RS content of di-starch phosphate was increased with increasing pH of the reaction mixture. X-ray patterns of RS from native and phosphorylated sago starches indicated that a crystalline structure transformation of those starches took place during preparation of RS.
PDF Abstract XML References Citation
Received: October 08, 2017;
Accepted: February 12, 2018;
Published: March 15, 2018
Copyright: © 2018. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Febby J. Polnaya, Djagal W. Marseno and Muhammad N. Cahyanto, 2018. Physical Properties and Digestibility of Resistant Starch from Phosphorylated Sago Starches. Pakistan Journal of Nutrition, 17: 199-206.
DOI: 10.3923/pjn.2018.199.206
URL: https://scialert.net/abstract/?doi=pjn.2018.199.206
DOI: 10.3923/pjn.2018.199.206
URL: https://scialert.net/abstract/?doi=pjn.2018.199.206
INTRODUCTION
Resistant starch (RS) is defined as the sum of starch and starch degradation products not absorbed in the small intestine of healthy individuals1. The RS can be found in processed food and raw food materials and is subdivided into four categories depending on the cause of resistance: RS1, physically located within the food matrix and are thus inaccessible to enzymes in the digestive tract; RS2, raw starch granules with associated crystallinity; RS3, retrograded starches formed when crystalline structures are formed and RS4, chemically modified (including cross-linked) starch2-4. Among the four types, RS4 or chemically modified starches are known to be the most resistant when treated with appropriate enzymes in vitro5.
The RSs are of particular interest because of their potential health benefits for humans. The RS is recognized as a type of nondigestible carbohydrate in humans, where it has been shown to be a mild laxative. It may almost totally ferment in the colon to short-chain fatty acids and it tends to reduce fecal pH and secondary bile acids6. Sang and Seib7 reported that the proportion of RS generated in the phosphorylated form increased as the level of the phosphorylating agent (STMP/STPP) increased. These data suggest that it may be desirable to increase the level of RS in foods through the modification of native starches by chemical methods. Mun and Shin5 reported that the addition of RS4, manufactured by chemical modification, can increase the RS level in food. Phosphorylation or cross-linking inhibits the migration of the molecules into a combining site with α-amylase.
Most research on RS has focused on starches from wheat, rice, maize, potatoes, peas, waxy maize and amylomaize3,4,8-10. The RS formation of starches derived from crops indigenous of the tropical regions, such as sago starch, was still limited. In nature, native sago starch contains chemically bound phosphorus at levels <0.01%11-13. The phosphate groups affect the properties and behavior of the sago starch11-13. To obtain higher phosphorus content, starch may be chemically phosphorylated.
Sago starch granules have a broad size range, between 20 and 40 mm with an oval or polygonal-shape and with a number of truncated oval granules14,15. Haska and Ohta16 found that sago starch is resistant to enzymatic degradation. Govindasamy et al.17 suggested a number of factors may well be responsible for the low levels of amylolysis of sago starch granules: (1) The α-amylase may be unable to attain access to the granular components. Enzymes are restricted by the specific molecular orientation of the starch chain components at the granular surface, the extensive degree of crystallinity, tight packing or the presence of minor components and (2) There may be limited surface available for absorption. This may reflect the comparatively small size of the granule or the fact that only a few suitable chain segments are available because this enzyme hydrolyzes only α(1→4) linkages that are remote from both the chain end and the branch point.
The interest of nutritionists and the food industry in phosphorylating and cross-linking starches is increasing and has led to an extensive investigation of the contribution of these starches to the nondigestible carbohydrate component of the diet and its associated physiological implications5. The extent of in vitro resistance of RS is related to the degree and type of modification. Increasing the degree of substitution of the starch may be expected to inhibit the entrance of enzyme molecules. Previous studies have demonstrated the properties of sago starch modified using STPP or POCl3 reagents, resulting in potentially starch resistant molecules13,18. Earlier researchers reported that the level of cross-linking made starch difficult to swell and gelatinize, so the pasting viscosity was very low13. Therefore, the objective of this study was to investigate the effects of the reactions of sago starch with STPP or POCl3 at pH 8, 9, 10 and 11 on the physical properties and in vitro digestibility of RS.
MATERIALS AND METHODS
Materials: The native sago starch (NSS) was donated by PT, Tiga Pilar Sejahtera Food Tbk., Indonesia. The starch was phosphorylated with sodium tripolyphosphate (STPP) or phosphorous oxychloride (POCl3). The moisture content of the sago starch ranged from 12-13%. The POCl3 was obtained from Merck (Germany), while food-grade STPP was obtained from the local market. Thermally stable α-amylase (63 U mL–1) and glucoamylase (151 U mL–1) for the resistant starch assay for the determination of the RS content were donated by the Sumber Manis factory (Pati, Center of Java). All chemicals used for analyses were of analytical grade.
Preparation of chemically modified starches: The STPP-treated sago starch was prepared using the methodology of Lim and Seib19. Fifteen grams of STPP was dissolved in 300 mL of water containing 15 g of Na2SO4. The pH of the solution was adjusted between 8 and 11 by adding 10% NaOH (Merck, Germany). The NSS (300 g) was dispersed in the solution. Then, the pH of the dispersion was readjusted with 5% NaOH and the total weight was brought to 667 g by adding water. The slurry was stirred for 1 h at 27°C and dried to 10-15% moisture at 40°C in a forced-air oven. To effect phosphorylation, the dried starch cake was heated for 2 h at 130°C in a forced-convection oven (Memmert, Germany). After being cooled to room temperature, the reaction mixture was dispersed in distilled water (350 mL). The starch was recovered by centrifugation (3,500 rpm, 10 min) and redispersed in 600 mL of distilled water. The pH of the dispersion was adjusted to 6.5. The starch was washed with water (4×600 mL) and dried overnight at 40°C in a cabinet dryer (CDR-9, China).
The POCl3-treated sago starch was prepared using the methodology of Felton and Schopmeyer20. The NSS (50 g, dry basis) was phosphorylated at various pH levels (from 8-11) with 4% POCl3 in the presence of 7.5 g of Na2SO4. POCl3 was added dropwise over a 20 min period, while maintaining the pH with 1.0 M NaOH. The starch dispersion was stirred at 25°C for 1 h and then adjusted to pH 6.5 with 1.0 M HCl (Merck, Germany). Modified starch was recovered by centrifugation (3,500 rpm, 10 min), washed with water (4×100 mL) and dried overnight at 40°C in a cabinet dryer.
Determination of functional groups: The functional groups of modified starches were characterized using an IR Prestige-21 FT-IR spectrometer (Shimadzu, Japan) using the KBr pellet technique. The equipment was operated with a scanning range from 4000-400 cm–1 21.
Determination of relative degree of cross-linking: The relative degree of cross-linking of the cross-linked starches was determined from the viscosity values, according to the method of Kaur et al.22. The peak viscosity of the starch samples was recorded using a Rapid Visco Analyser (Model RVA-4SA, Newport Scientific Pty Ltd. Warriewood, Australia). The starch slurry was heated from 50-95°C and was then held at 95°C for 2 min and 30 sec. Afterwards, the paste was cooled to 50°C and finally kept at 50°C for 2 min. The relative degree of cross-linking was calculated as follows22:
![]()
where, A is the peak viscosity in rapid viscosity units of the native starch sample and B is the peak viscosity of the phosphorylated starches.
X-ray diffraction measurements: The X-ray diffraction analysis was performed using an X-ray diffractometer (Lab X 116 XRD-6000, Shimadzu Japan) with a copper anode X-ray tube (Cu-Kα radiation) as described by Nara and Komiya23. The starch powders were packed tightly into sample holders. Each sample was exposed to the X-ray beam at 30 kV and 30 mA. The scanning region of the diffraction angle ranged from 2θ = 5°-30° in increments 0.40 with a count time of 1.0 sec, the rotary speed of the sample holder was 30 min–1. A smooth curve, that connected peak baselines, was computer-plotted on the diffractograms (Fig. 1). The area above the smooth curve was taken as the crystalline portion and the lower area between the smooth curve and the linear baseline which covered the 2θ range from 5°-35° was taken as the amorphous section. The ratio of the upper area to total diffraction was used as the degree of crystallinity. The equation for calculating the degree of crystallinity is as follows (Nara and Komiya23):
![]()
where, Ac is the crystalline area on the X-ray diffractogram and Am is the amorphous area on the X-ray diffractogram.
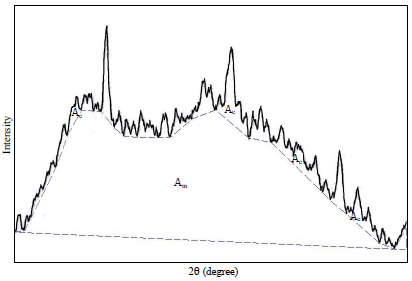 | |
| Fig. 1: | Measurement degree of crystallinity using UTHSCSA image tool software |
| Ac: Crystallinity region, Am: Amorphous region | |
Preparation and determination of resistant starch: The RS from native, mono- and di-starch phosphate were prepared according to the enzymatic-gravimetric method24, with slight modifications. The starch sample (4 g) was suspended in 160 mL of 0.08 M phosphate buffer (pH 5.5) (Merck, Germany). The solution was gelatinized in boiling water for 15 min, then cooled to 85°C. Forty microliters of α-amylase was added to the starch suspension and incubated at 85°C in a shaking water bath for 30 min. After cooling, the pH of the digested starch was adjusted to 4.5 by adding 1 N HCl (Merck, Germany). The digested starch was further hydrolyzed with 80 μL of glucoamylase and incubated for 80 min at 60°C in a shaking water bath. After incubation, the hydrolysate was heated at 100°C for 5 min, then centrifuged at 3,500 rpm for 10 min. The soluble starch fraction was precipitated with 95% ethanol (Sigma-Aldrich, Germany) to make an 80% concentration of alcohol and allowed to stand for over 1 h at room temperature. The residue was centrifuged at 3,500 rpm for 10 min. The RS precipitate was washed using 78% ethanol and subsequently centrifuged at 3,500 rpm for 10 min and then freeze-dried (Alpha 1-2 LD Plus, Germany).
To determine the resistant starch content, the hydrolyzed starch fraction was incubated with 10 mL of glucose oxidase/peroxidase reagent for 20 min at 20°C to determine the glucose content. Absorbance was measured using a UV-Vis 1650 spectrophotometer (Shimadzu, Japan) at 510 nm. The concentration of RS was calculated as follows:
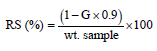
where, wt. sample is the initial weight (g) and G is the weight of glucose (g).
Statistical analysis: Data were statistically analyzed by one-way analysis of variance, with three replicates and significant differences were identified by Tukey’s test (p<0.05) using SAS 9.0 software (SAS, Inc.).
RESULTS AND DISCUSSION
FT-IR measurements: The FT-IR spectra of RS from NSS, mono-starch phosphate and di-starch phosphate are shown in Fig. 2. The FT-IR spectroscopy was used to verify the presence of phosphate groups in RS with chemical structures resulting from the hydrolysis of starches. Spectra were observed in the regions at 1172 and 956 cm–1, which originated from the P=O stretching vibration (ν P=O) and C-O-P (δ C-O-P), respectively. Heinze et al.25 showed, for the phosphate group in particular, that ν P=O is between 1150 and 1185 cm–1 while δ C-O-P is between 1055-950 cm–1.
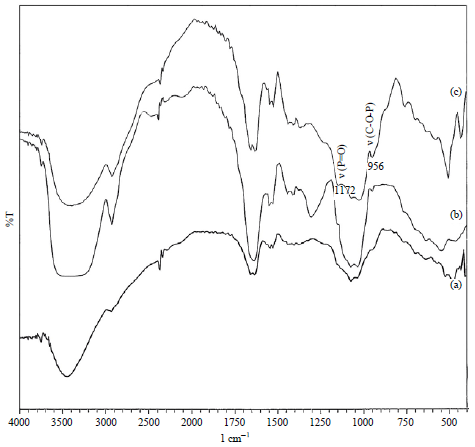 | |
| Fig. 2: | Fourier transform-infrared spectrum RS of (a) Native sago starch, (b) Mono-starch phosphate and (c) Di-starch phosphate |
This observation was also supported by the report of Polnaya et al.13, who showed that 31P NMR spectrum of starch phosphate has signals for mono-starch phosphate at δ 3.41 and 4.94 ppm and di-starch phosphate at the range δ -0.51 to 0.20 ppm. The FT-IR spectra of RS showed two uniform groups for ν P=O and δ C-O-P. The similarities in the spectra of RS are probably due to the hydrolysate of the RS preparation containing α,γ-dextrin enriched with phosphate groups.
Relative degree of cross-linking: The relative degree of cross-linking can be estimated by using RVA. From these results, it was found that di-starch phosphate treated at various pH levels (from 9-11) had different relative degrees of cross-linking ranged between 22.83 and 85.33 (Table 1). On the other hand, researchers were not able to estimate the relative degree of cross-linking in mono-starch phosphate treated at various pH levels (from 8-11) and POCl3-treated sago starch at pH 8 because of the relatively higher peak viscosity compared to NSS. Polnaya et al.13 showed that NSS phosphorylated with POCl3 produced predominantly di-starch phosphate and much less cyclic mono-starch phosphate in modified starch.
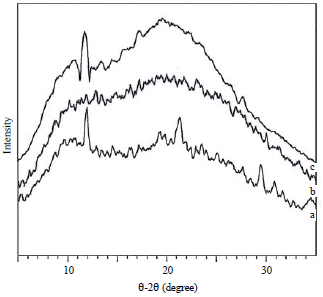 | |
| Fig. 3: | X-ray diffraction patterns of the (a) Native sago starch, (b) Mono-starch phosphate and (c) Di-starch phosphate |
These results indicated that the mono-starch phosphate was easier to paste because it contained more hydrophilic groups than NSS, while di-starch phosphate generated cross-linking, which reinforced the intermolecular linkages in the starch granule, resulting in more difficulty with pasting26. When NSS was treated with POCl3 in this experiment, researchers observed that pH 8 was not optimal to form di-starch phosphate. Therefore, mono-starch phosphate was formed predominantly. Cross-linking of starch molecules to give a di-starch phosphate was favored by alkalinity above pH 10 in the presence of a neutral sodium salt. This was expected because a higher pH catalyses starch phosphorylation13. Otherwise, mono-starch phosphates are formed13,27,28. At the high relative degree of cross-linking of sago starch, the peak viscosity of di-starch phosphate was lower than the NSS. The relative degree of cross-linking was great enough to restrict the swelling of the granules13.
Crystallinity of resistant starch: The X-ray diffraction patterns of RS from NSS, mono- and di-starch phosphate are shown in Fig. 3 and Table 2. The RS from mono- and di-starch phosphate showed a different pattern and degree of crystallinity than that of RS from NSS (Fig. 3), indicating that with gelatinization occurring in the boiling water prior to the enzyme hydrolysis process, the crystalline structure of RS from starches was destroyed and the degree of crystallinity of RS from starch phosphate was decreased compared to NSS.
| Table 1: | Relative degree of cross-linking and resistant starch content (%) of native sago starch, STPP and POCl3 treated sago starches with different pH reaction |
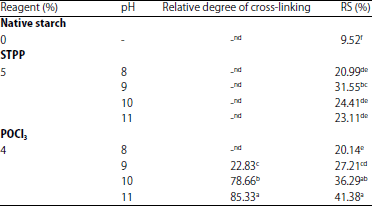 | |
Means superscripted with the same letters in the same column are not significantly different at p<0.05 level by Tukey’s test. ndRelative degree of cross-linking was not detectable | |
| Table 2: | X-ray diffraction intensities of peak and crystallinity of native sago starch, STPP treated sago starch at pH 9 and POCl3 treated sago starch at pH 11 |
 | |
| Means superscripted with the same letters are not significantly different at p<0.05 level by Tukey’s test | |
Mangala and Tharanathan29 showed different X-ray patterns among rice starch and its RS. With a higher degree of crystallinity (15.58%) (Table 2), RS from NSS contains mainly crystals of retrograded amylose and these structures resist enzyme hydrolysis, rendering a highly resistant material.
The RS from NSS exhibited peaks at 2θ = 9°, 11° and 21° (Fig. 3a), while the diffraction patterns of RS phosphate were indicated by the pronounced peaks at 2θ = 8°, 9°, 10° and 11° (Fig. 3b and c, Table 1). These peaks are not associated with any known starch crystalline structure. Polnaya et al.13 showed that NSS was characterized by a weak diffraction peak at 2θ = 5.67° and broad peaks at 2θ = 15.30°, 17.12°, 18.08° and 23.46°, which indicated the type C starch. Native and sago starch phosphate showed similar type and degree of crystallinity.
The RS phosphate exhibited lower crystallinity (~6%) than starch phosphate. Polnaya et al.13 showed that the mono- and di-starch phosphate had degrees of crystallinity ranging from 22.96-24.76%. The lower crystallinity of RS phosphate could be attributed to a poorly organized crystalline structure. The decrease in crystallinity of RS phosphate suggests that the disruption of the crystalline regions occurred during the gelatinization and hydrolysis processes. Alpha-amylase and glucoamylase hydrolysis can simultaneously solubilize both amorphous and crystalline regions of starch granules30. This is based on the observation that α-amylolysis did not produce an increase in crystallinity30. Crystallinity of barley starch has been shown to decrease during the later stages of α-amylolysis31. From these results, it was suggested that the substitution of phosphate groups might help increase the RS content from mono- and distarch phosphate compared to the crystallinity.
Resistant starch content: The RS content of NSS, mono-starch phosphate and di-starch phosphate measured by the RS assay procedure are shown in Table 1. The RS contents of NSS are less than 10%. The digestibility of NSS has often been related to crystallinity and is also partly affected by the presence of minor components, such as phosphate groups. Muhammad et al.11 and Polnaya et al.18 showed that the NSS contained <0.01% phosphorus. It has been reported that NSS is resistant to enzyme attack16 and is in general resistant to both microbial and enzyme digestion12.
The RS contents of mono- (20.99-31.55%) and di-starch phosphate (20.14-41.38%) were higher than that of NSS (9.52%). This was likely due to the introduction of phosphate groups to the starch molecules that contribute to the retardation of the enzyme-substrate complex. Chemical modification of starches, including esterification, etherification and cross-linking, renders α-(1-4) and α-(1-6) bonds resistant to hydrolysis32,33. Chemical modification has long been known to inhibit the in vitro digestibility of starch3. According to Abe et al.34, it is clear that the phosphate groups of starch prevent enzyme action and the large phosphorylated limit-dextrin molecules remain in the hydrolysate.
The di-starch phosphate with relative degrees of cross-linking >78% had higher RS content than that of the mono-starch phosphate (Table 1). These data show that the di-starch phosphate was more resistant to hydrolysis than the mono-starch phosphate. This may be the reason why di-starch phosphate showed higher RS content compared to NSS and mono-starch phosphate. These data are in accordance with the study of Woo and Seib3, who showed that cross-linking of starch with STPP imparts resistance to α-amylase hydrolysis (thus creating ‘RS’). Therefore, di-starch phosphate would be difficult to hydrolyze by amylolytic enzymes.
Starch phosphorylation using STPP was conducted at pH 8-11, with the reaction at pH 9 giving the highest RS content. This might have been due to the reaction at pH 9 giving the highest degree of substitution. The RS content of di-starch phosphate had the tendency to increase with the increase in pH. This was expected because a higher pH catalyzes a higher degree of cross-linking of starch. Increasing the degree of substitution or cross-linking also inhibits access of the enzyme. Song et al.10 showed that the level of di-starch phosphate was positively correlated with the level of RS4. Many studies have reported that the cross-linking reaction increases the RS level and is highly dependent on the degree of chemical modification3,9.
CONCLUSION
The RS was prepared from sago starch by phosphorylation with STPP or POCl3 at pH 8-11. The RS from NSS, mono- and distarch phosphate spectra, using FT-IR spectroscopy, were characterized by the presence of the phosphorus groups in the structure. It has been demonstrated that the content of RS could be improved greatly with phosphorylation. The highest RS content could be obtained by di-starch phosphate. After phosphorylation, the RS content was higher than that of NSS. The RS content of di-starch phosphate increased with increasing pH of the reaction mixture. The X-ray patterns of RS from native and phosphorylated sago starches indicated that a crystalline structure transformation of those starches took place during preparation of RS.
SIGNIFICANCE STATEMENT
This study reveals that phosphorylation of NSS can have beneficial effects on the structure of RS. It has also shown that the RS contents of mono- and distarch phosphate are higher than that of NSS. The results of this study will help other researchers in the formulation of new products, such as prebiotics, that can enhance human life.
REFERENCES
- Asp, N.G., 1992. Resistant starch. Proceedings for the 2nd plenary meeting of EURESTA: European FLAIR Concerted Action No. 11 on physiological implications of the consumption of resistant starch in man. Crete, 29 May-2 June 1991. Eur. J. Clin. Nutr., 46: S1-S148.
PubMedDirect Link - Brouns, F., B. Kettlitz and E. Arrigoni, 2002. Resistant starch and the butyrate revolution. Trends Food Sci. Technol., 3: 251-261.
CrossRefDirect Link - Woo, K.S. and P.A. Seib, 2002. Cross-linked resistant starch: Preparation and properties. Cereal Chem., 79: 819-825.
CrossRefDirect Link - Themeier, H., J. Hollmann, U. Neese and M.G. Lindhauer, 2005. Structural and morphological factors influencing the quantification of resistant starch II in starches of different botanical origin. Carbohydr. Polym., 61: 72-79.
CrossRefDirect Link - Mun, S.H. and M. Shin, 2006. Mild hydrolysis of resistant starch from maize. Food Chem., 96: 115-121.
CrossRefDirect Link - Topping, D.L. and P.M. Clifton, 2001. Short chain fatty acids and human colonic function: Roles of resistant starch and non starch polysaccharides. Physiol. Rev., 8: 1031-1064.
CrossRefPubMedDirect Link - Sang, Y. and P.A. Seib, 2006. Resistant starches from amylose mutants of corn by simultaneous heat-moisture treatment and phosphorylation. Carbohydr. Polym., 63: 167-175.
CrossRefDirect Link - Sievert, D. and Y. Pomeranz, 1989. Enzyme-resistant starch. I. Characterization and evaluation by enzymatic, thermoanalytical and microscopic methods. Cereal Chem., 66: 342-347.
Direct Link - Sang, Y., P.A. Seib, A.I. Herrera, O. Prakash and Y.C. Shi, 2010. Effects of alkaline treatment on the structure of phosphorylated wheat starch and its digestibility. Food Chem., 118: 323-327.
CrossRefDirect Link - Song, J.Y., J.H. Park and M. Shin, 2011. The effects of annealing and acid hydrolysis on resistant starch level and the properties of cross‐linked RS4 rice starch. Starch‐Starke, 63: 147-153.
CrossRefDirect Link - Muhammad, K., F. Hussin, Y.C. Man, H.M. Ghazali and J.F. Kennedy, 2000. Effect of pH on phosphorylation of sago starch. Carbohydr. Polym., 42: 85-90.
CrossRefDirect Link - Srichuwong, S., T.C. Sunarti, T. Mishima, N. Isono and M. Hisamatsu, 2005. Starches from different botanical sources I: Contribution of amylopectin fine structure to thermal properties and enzyme digestibility. Carbohydr. Polym., 60: 529-538.
CrossRefDirect Link - Polnaya, F.J., Haryadi, D.W. Marseno and M.N. Cahyanto, 2013. Effects of phosphorylation and cross-linking on the pasting properties and molecular structure of sago starch. Int. Food Res. J., 20: 1609-1615.
Direct Link - Ahmad, F.B., P.A. Williams, J.L. Doublier, S. Durand and A. Buleon, 1999. Physico-chemical characterisation of sago starch. Carbohydr. Polym., 38: 361-370.
CrossRefDirect Link - Yiu, P.H., S.L. Loh, A. Rajan, S.C. Wong and C.F.J. Bong, 2008. Physiochemical properties of sago starch modified by acid treatment in alcohol. Am. J. Applied Sci., 5: 307-311.
Direct Link - Haska, N. and Y. Ohta, 1992. Mechanism of hydrolysis of the treated sago starch granules by raw starch digesting amylase from Penicillium brunneum. Starch‐Starke, 44: 25-28.
CrossRefDirect Link - Govindasamy, S., C.G. Oates and H.A. Wong, 1992. Characterization of changes of sago starch components during hydrolysis by a thermostable alpha-amylase. Carbohydr. Polym., 18: 89-100.
CrossRefDirect Link - Polnaya, F.J., Haryadi, D.W. Marseno and M.N. Cahyanto, 2012. Preparation and properties of phosphorylated Sago starches. Sago Palm, 20: 3-11.
Direct Link - Lim, S. and P.A. Seib, 1993. Preparation and pasting properties of wheat and corn starch phosphates. Cereal Chem., 70: 137-144.
Direct Link - Singh, N., D. Chawla and J. Singh, 2004. Influence of acetic anhydride on physicochemical, morphological and thermal properties of corn and potato starch. Food Chem., 86: 601-608.
CrossRefDirect Link - Kaur, L., J. Singh and N. Singh, 2006. Effect of cross‐linking on some properties of potato (Solanum tuberosum L.) starches. J. Sci. Food Agric., 86: 1945-1954.
CrossRefDirect Link - Nara, S. and T. Komiya, 1983. Studies on the relationship between water-satured state and crystallinity by the diffraction method for moistened potato starch. Starch Starke, 35: 407-410.
CrossRefDirect Link - AOAC., 1990. Official Methods of Analysis. 15th Edn., Association of Official Analytical Chemists, Washington, DC., USA., Pages: 684.
Direct Link - Heinze, U., D. Klemm, E. Unger and F. Pieschel, 2003. New starch phosphate carbamides of high swelling ability: Synthesis and characterization. Starch‐Starke, 55: 55-60.
CrossRefDirect Link - Choi, S.G. and W.L. Kerr, 2004. Swelling characteristics of native and chemically modified wheat starches as a function of heating temperature and time. Starch‐Starke, 56: 181-189.
CrossRefDirect Link - Kasemsuwan, T. and J. Jane, 1994. Location of amylose in normal starch granules. II. Locations of phosphodiester cross-linking revealed by phosphorus-31 nuclear magnetic resonance. Cereal Chem., 71: 282-286.
Direct Link - Singh, J., L. Kaur and O.J. McCarthy, 2007. Factors influencing the physico-chemical, morphological, thermal and rheological properties of some chemically modified starches for food applications-A review. Food Hydrocolloids, 21: 1-22.
CrossRefDirect Link - Mangala, S.L. and R.N. Tharanathan, 1999. Structural studies of resistant starch derived from processed (autoclaved) rice. Eur. Food Res. Technol., 209: 38-42.
CrossRefDirect Link - Zhou, Y., R. Hoover and Q. Liu, 2004. Relationship between α-amylase degradation and the structure and physicochemical properties of legume starches. Carbohydr. Polym., 57: 299-317.
CrossRefDirect Link - Lauro, M., P.M. Forssell, M.T. Suortti, S.H.D. Hulleman and K.S. Poutanen, 1999. α-Amylolysis of large Barley starch granules. Cereal Chem., 76: 925-930.
CrossRefDirect Link - Hood, L.F. and V.G. Arneson, 1976. In vitro digestibility of hydroxypropyl distarch phosphate and unmodified tapioca starch. Cereal Chem., 53: 282-290.
Direct Link - Abe, J.I., Y. Takeda and S. Hizukuri, 1982. Action of glucoamylase from Aspergillus niger on phosphorylated substrate. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol., 703: 26-33.
CrossRefDirect Link