
Tooba Mirzapour
Institute of Bioscience, Putra University, Malaysia, Selangor, Malaysia
Mansoureh Movahedin
Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University, Tehran, Iran
Tengku Azmi Bin Tengku Ibrahim
Institute of Bioscience, Putra University, Malaysia, Selangor, Malaysia
Abd Wahid Haron
Institute of Bioscience, Putra University, Malaysia, Selangor, Malaysia
Zohreh Makoolati
Department of Anatomical Sciences, School of Medical Sciences, Tarbiat Modares University, Tehran, Iran
Mohamadreza Nowroozi
Department of Urology, Faculty of Medicine, Tehran University of Medical Science, Tehran, Iran
Journal of Biological Sciences
Year: 2010 | Volume: 10 | Issue: 8 | Page No.: 730-738







ABSTRACT
Exogenesis (cross-species) germ cell transplantation provides an opportunity to investigate fundamental aspects of spermatogenesis. In this study, testis biopsies of patients with maturation arrest of spermatogenesis during a one year ago were first minced mechanically into small pieces and then Spermatogonial Stem Cells (SSCs) and Sertoli cells isolated by the two- step enzymatic digestion, were plated and grown on DSA-Lectin coated dishes in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal calf serum. Transplantation of human spermatogonial cells into mouse recipient testis was performed on day 7 (before colony formation) and 2 weeks after culturing (colony formation). The effects of different concentrations of spermatogonial cell on quantity of transplantation and percent of colonized seminiferous tubules were assayed during 8 weeks after transplantation. The result showed that SSCs can be observed on the basement membrane of the seminiferous tubules in place of spermatogonial stem cells and proliferation occurs about 4 weeks after transplantation. The difference in donor cells concentration had more effect on colonization of mouse recipient testis (p<0.05). It will be an alternative approach for the repopulation of infertile seminiferous tubules and preservation of fertility, in the future.
PDF Abstract XML References Citation
Received: September 08, 2010;
Accepted: January 06, 2011;
Published: February 26, 2011
How to cite this article
Tooba Mirzapour, Mansoureh Movahedin, Tengku Azmi Bin Tengku Ibrahim, Abd Wahid Haron, Zohreh Makoolati and Mohamadreza Nowroozi, 2010. Effect of Donor Cells Concentration on Colonization of Human Spermatogonial Stem Cells in Recipient Mouse Testes. Journal of Biological Sciences, 10: 730-738.
DOI: 10.3923/jbs.2010.730.738
URL: https://scialert.net/abstract/?doi=jbs.2010.730.738
DOI: 10.3923/jbs.2010.730.738
URL: https://scialert.net/abstract/?doi=jbs.2010.730.738
INTRODUCTION
Spermatogenesis is a highly controlled procedure of cell proliferation and differentiation that starts at puberty (5-7 days after birth in rodents and 10-13 years after birth in men) and maintains during life (Dym et al., 2009). A few number of Spermatogonial Stem Cells (SSCs) that attached to the basement membrane of the seminiferous tubules are responsible for the beginning and preservation of spermatogenesis. The characterization of SSCs is ability to proliferate and self-renew extensively as well as to differentiate (Tehranipour, 2010) and ultimately form spermatozoa (Honaramooz et al., 2002; Schlatt et al., 2002).
Germ cells are developed in association with the Sertoli cells. These cells have nutrients, regulatory/paracrine growth factors and structural support for germ cells. The growth factors have an important role in providing an environment for cell growth, proliferation and differentiation which is affected in an organ-specific way by endocrine or paracrine mechanisms (Schlatt et al., 1997; Wahab-Wahlgren et al., 2003).
The cells that are lost during physiological turnover, or damaged by injury, aging and disease can replace by stem cells derived from mammalian adult tissues (Foroutan et al., 2010; Tadokoro et al., 2002). Several studies have suggested that spermatogonial stem cells diminish or lose their activity as male ages. A valuable form for studying the molecular and cellular biology of male germ cell is designing culture conditions that control the self-renewal vs. differentiation of Spermatogonial Stem Cells (SSCs) in vitro. It will allow the development of new therapeutic strategies for treatment of infertility. Based on previous studies, the potential damage of spermatogonial stem cells, with the possibility of infertility is a side effect of cancer treatment (Kubota and Brinster, 2006). In such cases for preservation of fertility it has been suggested that testicular biopsies be isolated prior to treatment and spermatogonial stem cells be proliferated in vitro and transplanted back into the patient’s testis (Kossack et al., 2008).
The transplantation technique provides unique opportunities for practical applications of spermatogonial stem cell, such as restoration of male fertility, germ-line preservation. Brinster and Zimmermann (1994) performed the pioneering work of germ cell transplantation in rodent models. Later studies showed that intratubular microinjection of germ cells suspension via the rete testis, restimulated spermatogenesis in spermatogonial stem cells and these cells divided and formed differentiating spermatogonia, entered meiosis divisions and ultimately formed spermatozoa (Nagano, 2002). This technique has been performed in primates, rodents, mice, macaque monkey, goat and cattle (Brinster and Nagano, 1998; Dirami et al., 1999; Radford et al., 1999; Russell et al., 1990; Schlatt et al., 1999)
The purpose of this research was to study the effects of different concentrations of human spermatogonial stem cells as donor cells on quality of colonized seminiferous tubules of recipient mouse testis. For this purpose, isolated hSSCs from testis of azoospermia patients were co-cultured on Sertoli cells as a feeder layer in vitro system. Then spermatogonial-cell-derived colonies were isolated and different concentrations of hSSCs transplanted into the seminiferous tubules (8 testes in total) of busulfan-treated immunodeficient mice and quality of transplantation was assayed.
MATERIALS AND METHODS
Sample collection: Human testis biopsies were routinely obtained from the diagnosis of maturation arrest of spermatogenesis through the clinical practice of Imam Khomeini Hospital (Tehran, Iran) patients (n = 8 , aged 28-50 years, average 35.6 years). The use of human testicular biopsy sample and the experimental protocol were approved by National Research Council guidelines of Tarbiat Modares University (Tehran, Iran).
Cell isolation: The testis biopsies from patients were placed into a medium and transferred to the laboratory within 60 min. They were minced into small pieces and suspended in Dulbecco’s Modified Eagle medium (DMEM; Gibco, Paisley, UK), which contained 0.5 mg mL-1 collagenase, 0.5 mg mL-1 trypsin, 0.5 mg mL-1 hyaloronidase and 0.05 mg mL-1 DNase, for 30 min (with shaking and a little pipetting) at 37°C. The obtained spermatogenic tubules and cells were then centrifuged in 1000 rpm at 4 min and resuspended in 2 mL of DMEM. They were washed three times in DMEM and removed most of the interstitial, fibroblast and endothelial cells. A second digestion step (45 min at 37°C) was performed by adding a fresh mixture of DMEM and enzymes to the seminiferous tubules component. After this treatment clumps of cells (A mixture of germ cells and Sertoli cells) were agitated gently with a Pasteur pipette for 4 min. The cells were centrifuged at 2200 rpm for 4 min at 37°C and isolated from the residual tubule fragments. The cells were filtered through a 70 μm nylon filter, washed twice and fresh medium was added. The Sertoli cells were isolated using a modified version of the method described by Scarpino et al. (1998). Briefly coated plastic dishes (with a diameter of 60 mm) were organized by incubation with a solution of 5 μg mL-1 of Datura Stramonium Agglutinin (DSA; Sigma) in PBS at 37°C for 60 min. They were washed three times with DMEM containing 0.5% Bovine Serum Albumin (BSA; Sigma).
The mixed suspension of the cells obtained by two-step enzymatic digestions at a concentration of 3.5x105 cells cm-2 was located on lectin-coated dishes and incubated at 32°C in 5% CO2 in air. After 2-3 h of incubation, the non-adhering cells were collected by twice washing with the medium.
Purified Sertoli cells were proliferated and enhanced approximately after 72 h. These cells were detached by ethylenediamine tetraacetic acid (EDTA)-trypsin treatment (0.02% EDTA-0.1% trypsin in Ca2+- and Mg-free PBS) for 5 min at 37°C. Then they were counted and used to preferred concentrations into each well of a 12-well multi dish (2x105 cells cm-2). In the presence of 10% fetal bovine serum they were cultured in DMEM at 32°C (FBS; Gibco). By this method the Sertoli cells were isolated with more than 95% purity.
Co-culture of SSCs on sertoli cells and morphological evaluation of colonies: In this study, Sertoli cells were used as feeder layer for the culture of spermatogonial stem cells individually. Sertoli cells formed a confluent layer, three-four days after incubation of these cells on lectin-coated wells. Then spermatogonial cells (20x103 cells cm-2) were co-cultured on top of Sertoli cells for 2 weeks. Every three days the medium was changed and replaced with fresh medium. A hemaocytometer was used for determining the number of the Sertoli and spermatogonial cells for co-culture. Dye exclusion test (0.04% trypan blue solution) was done for evaluation of cell viability.
Identity confirmation of sertoli and spermatogonial cells Alkaline phosphatase reactivity: In previous studies, alkaline phosphatase reaction has been described as a marker for embryonic stem cells and spermatogonial stem cells (Guan et al., 2006; Kubota et al., 2004; Nagano et al., 2001). For alkaline phosphatase assay, the spermatogonial-cell-derived colonies and Sertoli cells were fixed for 10 min in a solution containing 2.5 mL citrate, 6.5 mL acetone and 0.6 mL formaldehyde in distilled water and incubated for 30 min in a solution containing 0.5 mg mL-1 Fast Red Violet (Sigma) and 40 μL mL-1 α-naphtol phosphate (0.25% solution). After rinsing in water, the slides were stained with haematoxilin and after dehydration by alcohol they were mounted and observed using light microscopy. The positive control group was brush border of villous from mouse intestine. For positive control group, the sections of neonatal mouse intestine were first deparaffinised, rehydrated and then done alkaline phosphatase assessment following mentioned protocol for SSCs colonies.
Immunocytochemical evaluation: Vimentin, which is a molecular marker for Sertoli cells, was detected in the feeder monolayer cells by immunocytochemistry technique. For this purpose, the cells grown on the glass slides were fixed in 4% paraformaldehyde at room temperature for 20 min. Then the slides were rinsed with PBS. Following permeabilization by 0.2% Triton X-100 (MPbiomed ICN) for 1 h which facilitates antibody penetration the slides were washed with PBS supplemented with 0.2% BSA. Non-specific antigens were blocked with 10% normal goat serum (Vector, Burlingame, CA, USA). The slides were then incubated for overnight at 37°C with a mouse monoclonal anti-vimentin antibody (diluted 1: 200; sigma, USA). This antibody is a marker for Sertoli cells (Anway et al., 2003). After extensively washing with PBS, the second antibody (goat anti-mouse IgM labelled with fluorescein isothiocyanate (FITC); diluted 1:100, Sigma, USA) was added for 2 h. Control staining consisted of the same process excluding reaction against Vimentin and anti-Vimentin.
Recipient mice testis and human spermatogonial cells transplantation: Male mature (6-8 weeks of age; n = 15) National Medical Research Institute mice, from original stocks of Razi Laboratory (Tehran, Iran), were maintained with free access to food and water at the Animal Facilities of Tarbiat Modares University (Tehran, Iran). A total of 8 human donor cell samples were prepared from testicular biopsy specimens obtained from 8 patients, proliferated in culture system and transplanted into the seminiferous tubules (15 testes in total) of busulfan-treated immunodeficient mice. Suspension of the donor spermatogonial cells in 10-15 μL DMEM were injected into the seminiferous tubules in the left testis of each recipient mouse via rete testis. Right testis was considered as control groups.
To destroy endogenous spermatogenesis, the recipient mice were treated at 6-8 weeks of age with busulfan (20 mg kg-1 i.p.; Sigma) at least 4 weeks before donor cell transplantation (Nagano et al., 2002). These animals were used as recipients of human spermatogonial cells transplantation.
Transplantation was performed by obtained cells from three different processes of culture, on day 0 (isolated testicular cells before culturing), day 7 (one week after culture) and day 14 (two weeks after culture). For evaluating of effects of donor cell concentration on percentage of colonized mouse seminiferous tubules, obtained human spermatogonial cells from this processes were transplanted into seminiferous tubules of the left testes in 3 groups including: the cells with a concentration of 8.5±2.1x106 mL (Mean±SEM) of SSCs as very low concentration, the cells with a concentration of 20.3±4.3x106 mL (Mean±SEM) of SSCs as low and the cells with a concentration of 41.2±5.3x106 mL (Mean±SEM) of SSCs as high concentration.
SSCs with very low concentration were testicular cells before colony formation while low and high concentrations of SSCs were related to colony-derived cells after culturing. Effects of donor cell concentration on percentage of colonized seminiferous tubules in recipient mouse testis (homing spermatogenesis) was shown as a fraction of colonized seminiferous tubules on injected seminiferous tubules (Table 1).
In vivo colonizing ability of transplanted cells in mouse testes after transplantation was evaluated by tracing of 5-bromo- 2-deoxyuridine (BrdU)-incorporated cells. For this purpose 0.1 mm BrdU was added to the culture medium (final concentration) 72 h before transplantation and co-cultured cells were incorporated with BrdU (Sigma). Then, the adult recipient mice were anesthetized using ketamine 10% and xylazine 2% (Alfasan,Woerden, The Netherlands) . The co-cultured cells were detached using EDTA-trypsin treatment for 5 min at 37°C and the Sertoli cells were isolated by DSA lectin immobilization. The remaining spermatogonia were collected and transplanted into the seminiferous tubules of the recipient mice.
Immunohistochemical detection of incorporated 5-bromo-2-deoxyuridine (BrdU) in proliferating spermatogonial cells after transplantation: Eight weeks after transplantation the control and transplanted mice testes were examined by immunohistochemistry. For this purpose after fixation of the testes tissue in 4% paraformaldehyde, they were dehyderated and embedded in paraffin. In order to visualize the donor-derived spermatogenesis the testis tissue were sectioned by microtome and immunostained with a primary anti-BrdU (Sigma).
For immunohistochemical staining, after deparaffining and rehydration of the sections, using xylene and a series of ethanol dilutions, they were washed twice in TBS buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl) for 5 min each followed by oven antigen retrieval at (60°C) for 120 min in 10 mM sodium citrate solution, pH 6.0 and then cooled in TBS.
| Table 1: | Percent of colonized seminiferous tubules of recipient mouse testis /total in different concentration of SSCs as donor cells |
 | |
| aSignificant difference. Very low concentration of SSCs in the same column (p<0.05) | |
The slides were placed into 2 M HCl for 30 min. Subsequently 0.1 m borate buffer (pH 8.5) was added to the slides and put at room temperature for 20 min. Following permeabilisation by 0.03% Triton X-100 (MPbiomed ICN) to facilitate antibody penetration, the cells were washed with PBS. Then a blocking solution containing 10% normal goat serum (Vector, Burlingame, CA, USA) was applied for the sections at 37°C for 30 min. After blocking, an aliquot of 50 μL mouse monoclonal anti-BrdU antibody (Sigma, 1:200 diluted in PBS containing 1% BSA) was added and the sections were incubated at 4°C overnight.
After the sections were extensively washed with PBS the second antibody (anti-mouse IgG; Sigma-Aldrich, Steinheim, Germany; conjugated with fluorescein isothiocyanate (FITC); diluted 1:100), was applied for 2 h at room temperature. The sections were washed in PBS again. Then they were dehydrated and mounted with entellane. The control slides had similar conditions apart from for the removal of the first antibody.
Statistical analysis: Values from samples receiving the same treatment were pooled for calculation of their means and standard errors (SEMs). One-way analysis of variance and Tukey-test were used to determine significant differences between the different experimental conditions. Probability values less than 0.05 were considered statistically significant.
RESULTS
Two different cell types with different sizes and morphology were obtained from the seminiferous tubules of human testes. The first type with 8.1-9.2 μm in diameter and an irregular outline appeared granular. These cells created a monolayer of cells after proliferation (Sertoli cells as feeder layer) (Fig. 1b), showing no Alkaline phosphatase reactivity (AP) (Fig. 2a, b). The second type which was spherical and larger than the first one, had a diameter of 15-17.2 μm with two or three eccentrically located nucleoli (Fig. 1a). These cells created a prominent colony after proliferation and passage during two weeks of culture (Fig. 1c, d). In other words, after culturing human spermatogonial stem cells on Sertoli cells many multicellular colonies of SSCs was formed. The diameter and number of colonies was increased during two weeks of culture.
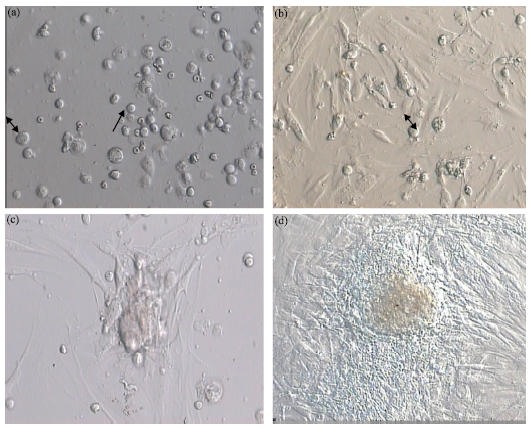 | |
| Fig. 1: | (a) Two different cell types with different sizes and morphology were obtained from the seminiferous tubules of human testes. The first type (↓) with 8.1-9.2 μm in diameter and an irregular outline appeared granular; (b) These cells created a monolayer of cells after proliferation (Sertoli cells as feeder layer). The second type ( |
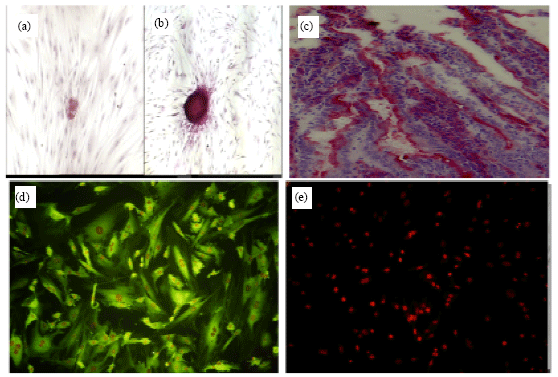 | |
| Fig. 2: | The cells in the colony of human spermatogonial cells was positive for alkaline phosphatase reactivity but monolayer of sertoli cells did not show any alkaline phosphatase reactivity (a and b); (c) Mouse intestine as positive control group for alkaline phosphatase reaction; (d) Vimentin immunocytochemistry was positive in Sertoli cells and (e) Control group for immunocytochemistry of vimentin. Magnification: x200 |
Similar to the control conditions, consisting of mouse intestine (brush border of villous) that showed Alkaline Phosphatase reactivity (Fig. 2c) this small germ cell clumps, also had AP activity clearly (Fig. 2a- e). Based on previous studies, alkaline phosphatase reaction has been described as a marker for spermatogonial stem cells (Guan et al., 2006; Kubota et al., 2004; Nagano et al., 2001). In our study, Vimentin that is the intermediate filament of mature human Sertoli cells was observed in culture system (Fig. 2d).
Colonizing ability of cultured human spermatogonial stem cells in mouse testes: To evaluate efficiency and colonizing ability of human spermatogonial stem cells in recipient mouse testis, a transplantation technique was performed. 5-Bromo-2-deoxyuridine was added 72 h before transplantation and staining was examined just before transplantation. The results showed that more than 95% of the total transplanted cells were labeled with BrdU (Fig. 3a, b). After transplantation, the cells showing nuclear BrdU staining (FITC) were considered as transplanted cells (Fig. 3c). Transplanted spermatogonial cells located on basement membrane of the seminiferous tubules of the recipient mouse testes (Fig. 3c). Right testis was considered a control group (Fig. 3d).
The results showed that the intercellular space found between Sertoli cell processes and germ cells was similar to that expected in a typical normal relationship of germ cells to Sertoli cells in active mouse testis (Fig. 3c).
In our study, donor human cells were observed as single or paired cells at 4 weeks after transplantation. This is a morphological characteristic typical of undifferentiated spermatogonia (Nagano et al., 2002). Extensive proliferation patterns with single or short chains of cells, which did not form a cluster, were observed on the basement membrane of seminiferous tubules in many recipient testes. These cells were found dispersed in the seminiferous tubules at 8 weeks after transplantation (Fig. 3c).
In comparison of different concentrations of transplanted cells in percentage of colonized seminiferous tubules (homing spermatogenesis), analysis of variance indicated no difference in colonization percentage between low and high concentration groups (p>0.07). But in concentration of 8.5x106 cell mL-1 (very low), the percentage of homing spermatogenesis was low and decreased rather than two other groups significantly (p<0.05).
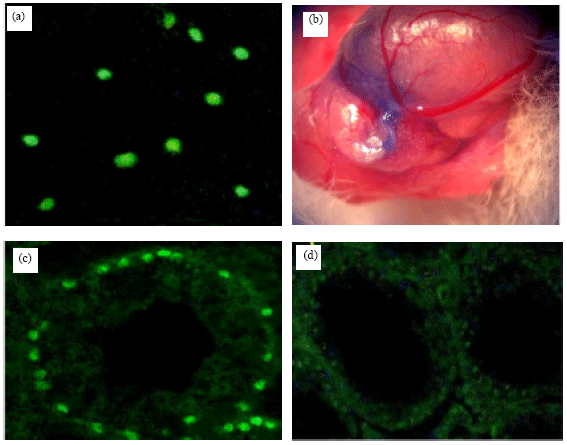 | |
| Fig. 3: | (a) 5-Bromo-2-deoxyuridine (BrdU) was added and staining was examined in cultured spermatogonial cells before transplantation. The cells with positive nuclear BrdU staining (fluorescein isothiocyanate) were as transplanted cells; (b) Transplanted human spermatogonial cells into rete testis. It was pretreated with busulfan to destroy endogenous spermatogenesis; (c) Donor human cells in mouse seminiferous tubule 8 weeks after transplantation. Most donor cells are present as single or paired cells and do not form a cluster and (d) The right testis was as the control group. They did not show any donor-derived spermatogenesis. Magnification: x200 (in a,b,d); x400 (in c) |
DISCUSSION
As we know, (a sentence has deleted) patients suffering from malignancies, chemotherapy or X-ray treatment may bring about permanent azoospermia or long azoospermic periods without recovery of spermatogenesis. In these patients, a potentially valuable technique offering gonadal protection fertility is germ cell transplantation. On the other hand, in oncological patients fertility can be maintained by reinitiating spermatogenesis from stem cells, which were transplanted into the seminiferous tubules (Schlatt et al., 1999).
Our findings demonstrated that short-term co-culturing of adult hSSCs on Sertoli cells influenced spermatogonial proliferation in vitro, so that high multicellular colonies formed in culture system in vitro. This is in agreement with study of Koruji et al. (2009) that obtained similar results in co-culture of adult mouse SSCs on Sertoli cells. Other studies have obtained similar results in culture of spermatogonial stem cells from other species (Van der Wee et al., 2001). So, in this study, sertoli cells were used as a feeder layer for culturing of adult human testicular cells from patients with maturation arrest in spermatogenesis.
In contrast to other feeder layers such as mouse embryonic fibroblast cells (MEF) or STO cells, the sertoli cells provide a befitting environment for the interaction of signaling networks that regulate the fate of spermatogonial stem cells (Hofmann et al., 2003; Izadyar et al., 2003; van der Wee et al., 2001). These cells do not have disadvantages of MEF, including a limitation of passage numbers that is optimal only between the 4th and 6th passages (Richards et al., 2004; Stojkovic et al., 2004). In other words, the use of feeder cells originating from the patient’s own testicular tissue will facilitate future clinical application importantly, because this modification avoids the use of animal products (Sadri-Ardekani et al., 2009) that could lead to viral infection and generate cells with tumorigenic potential (Stacey et al., 2006).
In this study for confirmation of Sertoli cells, the size, morphology and alkaline phosphatase activity were utilized as parameters for characterizations. In addition to that, specific marker detection was carried out using Vimentin immunocytochemistry. Feeder-monolayer of Sertoli cells did not show alkaline phosphatase activity and this finding is in agreement with that reported by Scarpino et al. (1998) who demonstrated the Sertoli cells do not exhibit alkaline phosphates activity. In this study Spermatogonial-derived colonies showed positive alkaline phosphatase activity. Vimentin is the intermediate filament of mature human Sertoli cells and in normal testes, expression of Vimentin has been demonstrated in Sertoli cells (Richburg and Boekelheide, 1996; Rogatsch et al., 1996). In this experiment, Vimentin immunocytochemistry was positive for Sertoli cells.
In the present investigation, functional and colonizing ability of human spermatogonial stem cells in mouse testes evaluated by transplantation technique. Furthermore, previous xenogeneic transplantation experiments using several animals species as donors and mice as recipients have indicated that the spermatogonial stem cells of many animal species can colonize mouse testes (Dym et al., 2009; McLean, 2005; Nagano et al., 2002, 2003; Schlatt et al., 1997). Rodent and primates diverged approximately 100 million years ago so, mouse seminiferous tubule somatic cells (e.g., Sertoli cells) can provide more adequate support for human spermatogonial stem cell survival and proliferation .
In this study, in concentration of 8.5±2.1x106 cell mL-1 of SSCs, the percentage of colonized seminiferous tubules (seminiferous tubules with homing spermatogenesis) was low and they were increased by exceeding of injected cells from very low concentration to low concentration (p<0.05) (Table 1). In fact, proliferation of human spermatogonial stem cells in co-culture system and preparation of a special environment for increasing the number of colony cells probably is an important factor to do successful homing spermatogenesis. It is in agreement with previous studies that have found identification of spermatogonial stem cell culture conditions for long-term culture, renewal and differentiation can be highly useful for the germ cell transplantation technique (Dirami et al., 1999). However, this findings showed a marked difference with study of Nagano et al. (2002) that reported the increase of donor cells concentration does not have any effect on percentage of colonized seminiferous tubules by human spermatogonia cells.
The comparison of the colonization percentage between two concentration of donor cells (low and high concentration groups) showed that increasing of donor cells to high concentration appeared to have more effect on the efficiency of colonization but observed no significant difference between them (p>0.07).
In this study it seems that the interactions between human spermatogonial cells and immunodeficiency mouse testis environment may be optimal and appropriate surface molecules and growth factors are available (McLean et al., 2001) because extensive proliferation patterns of human donor cells and regular location of them on the basement membrane of the seminiferous tubules was observed after transplantation. In fact, the Sertoli cell of mice supported the implanted human germ cells and identified that the interactions between germ and Sertoli cells are not necessarily species specific. This finding is in agreement with study of Dobrinski et al. (1999), Nagano et al. (2001) and Nagano et al. (2002) who demonstrated in the monkey and human testis. They mentioned that transplanted germ cells colonized the mouse testis, but spermatogenesis was incomplete and became arrested at the stage of spermatogonial division.Based on our study three different concentrations of SSCs did not significantly influence the human spermatogonial differentiation in mouse testes 8 weeks after transplantation. It is possible that exogenous environmental factors, derived from humans are required for maintenance of human spermatogenesis in mouse testes. Alternatively, removal of mouse Sertoli cells and replacement with human Sertoli cells or homologous transplantation of spermatogonial stem cells that obtained from a patient of azoospermia (maturation arrest) to seminiferous tubules of that patient individually after in vitro culture, may allow differentiation of transplanted stem cells. The efficient use of autologous stem cell transplantation in patients who may suffer destruction of germ cells as a result of irradiation or chemotherapy for cancer is valuable therapeutic tools for treatment but continued investigation of these approaches are needed in future (Watt and Hogan, 2000).
Since, time of spermatogenesis is controlled by the germ cell or Sertoli cell in xenogeneic transplants (Franca, 1998) and total spermatogenesis(from dark spermatogonia to sperm) in human take time 90 days and differentiated spermatogonia obtain after 27 days, in agreement with previous studies we used an average of 8 weeks period for assessment of transplantation efficiency in our study.
In many previous studies has been shown that development of spermatogonial transplantation technique and repopulation of sterile seminiferous tubules is a powerful tool for detecting of activity, study of biology and quantitative analyses of SSCs. In the future, germ cell transplantation might become an alternative way for the preservation of fertility (Schlatt et al., 1999) Similar to gonadal protection by pretreatment regimens before cancer therapy (Ward et al., 1990).
CONCLUSION
In this study we established a successful short-term culture of testiscular germ cells and the number of germ cells increased continuously during subcultures. Then, many colony-derived cells of human spermatogonial cells were transplanted into the seminiferous tubules of recipient mouse testis pretreated with busulfan. These cells survived on basement membrane of the seminiferous tubules in mouse testes for 8 weeks and proliferated during the first month after transplantation. But differentiating spermatogonia cells were not identified and meiotic differentiation was not observed in mouse testes.
ACKNOWLEDGMENTS
The authors appreciate current and previous members of the Tarbiat Modares university laboratory (Cell Culture Lab, Tehran, Iran) and University Putra Malaysia lab. (Cell Culture and Electron Microscopy Lab, UPM, Malaysia) for helpful discussions and technical support. I appreciate Dr. Movahedin who had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- Anway, M.D., J. Folmer, W.W. Wright and B.R. Zirkin, 2003. Isolation of Sertoli cells from adult rat testes: An approach to ex vivo studies of Sertoli cell function. Biol. Reprod., 68: 996-1002.
PubMed - Brinster, R.L. and M. Nagano, 1998. Spermatogonial stem cell transplantation, cryopreservation and culture. Semin. Cell Dev. Biol., 9: 401-409.
CrossRef - Brinster, R.L. and J.W. Zimmermann, 1994. Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA., 91: 11298-11302.
Direct Link - Dirami, G., N. Ravindranath, V. Pursel and M. Dym, 1999. Effects of stem cell factor and granulocyte macrophage-colony stimulating factor on survival of porcine type A spermatogonia cultured in KSOM. Biol. Reprod., 61: 225-230.
Direct Link - Dobrinski, I., M.R. Avarbock and R.L. Brinster, 1999. Transplantation of germ cells from rabbits and dogs into mouse testes. Biol. Reprod., 61: 1331-1339.
Direct Link - Dym, M., M. Kokkinaki and Z. He, 2009. Spermatogonial stem cells: Mouse and human comparisons. Birth Defectes Res., 87: 27-34.
PubMedDirect Link - Foroutan, T., A. Hosseini, A.A. Pourfatholah, M. Soleimani, K. Alimoghadam and N. Mosaffa, 2010. Peritoneal mesothelial progenitor or stem cell. J. Biol. Sci., 10: 460-464.
CrossRefDirect Link - Franca, L., T. Ogawa, M.R. Avarbock, R.L. Brinster and L.D. Russell, 1998. Germ cell genotype controls cell cycle during spermatogenesis in the rat. Biol. Reprod., 59: 1371-1377.
Direct Link - Guan, K., K. Nayernia, L.S. Maier, S. Wagner and R. Dressel et al., 2006. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature, 440: 1199-1203.
CrossRefDirect Link - Hofmann, M.C., K.S. Van Der Wee, J.L. Dargart, G. Dirami, L. Dettin and M. Dym, 2003. Establishment and characterization of neonatal mouse sertoli cell lines. J. Androl., 24: 120-130.
PubMedDirect Link - Honaramooz, A., S.O. Megee and I. Dobrinski, 2002. Germ cell transplantation in pigs. Biol. Reprod., 66: 21-28.
Direct Link - Izadyar, F., K. Den Ouden, L.B. Creemers, G. Posthuma, M. Parvinen and D.G. De Rooij, 2003. Proliferation and differentiation of bovine type A spermatogonia during long-term culture. Biol. Reprod., 68: 272-281.
PubMedDirect Link - Koruji, M., M. Movahedin, S.J. Mowla, H. Gourabi and A.J. Arfaee, 2009. Efficiency of adult mouse spermatogonial stem cell colony formation under several culture conditions. Cell. Dev. Biol. Anim., 45: 281-289.
CrossRefDirect Link - Kossack, N., J. Meneses, S. Shefi, H.N. Nguyen and S. Chavez et al., 2008. Isolation and characterization of pluripotent human spermatogonial stem cell-derived cells. Stem Cells, 27: 138-149.
PubMed - Kubota, H., M. Avarbock and R.L. Brinster, 2004. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA., 101: 16489-16494.
CrossRefDirect Link - Kubota, H. and R.L. Brinster, 2006. Technology insight: In vitro culture of spermatogonial stem cells and their potential therapeutic uses. Nat. Clin. Pract. Endocrinol. Metab., 2: 99-108.
PubMed - McLean, D.J., 2005. Spermatogonial stem cell transplantation and testicular function. Cell. Tissue Res., 322: 21-31.
CrossRefDirect Link - McLean, D.J., D.S. Johnston, L.D. Russell and M.D. Griswold, 2001. Germ cell transplantation and the study of testicular function. Trends Endocrinol. Metab., 12: 16-21.
PubMedDirect Link - Nagano, M., J.R. McCarrey and R.L. Brinster, 2001. Primate spermatogonial stem cells colonize mouse testes. Biol. Reprod., 64: 1409-1416.
PubMedDirect Link - Nagano, M., P. Patrizio and L. Brinster, 2002. Long-term survival of human spermatogonial stem cells in mouse testes. Fertil. Steril., 78: 1225-1233.
CrossRefPubMedDirect Link - Nagano, M., B.Y. Ryu, C.J. Brinster, M.R. Avarbock and R.L. Brinster, 2003. Maintenance of mouse male germ line stem cells in vitro. Biol. Reprod., 68: 2207-2214.
CrossRefDirect Link - Radford, J., S. Shalet and B. Lieberman, 1999. Fertility after treatment for cancer. Questions remain over ways of preserving ovarian and testicular tissue. BMJ., 319: 935-936.
PubMedDirect Link - Richards, M., S.P. Tan, J.H. Tan, W.K. Chan and A. Bongso, 2004. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem Cells, 22: 51-64.
PubMedDirect Link - Richburg, J.H. and K. Boekelheide, 1996. Mono-(2-ethylhexyl) phthalate rapidly alters both sertoli cell vimentin filaments and germ cell apoptosis in young rat testes. Toxicol. Applied Pharmacol., 137: 42-50.
PubMedDirect Link - Rogatsch, H., D. Jezek, A. Hittmair, G. Mikuz and H. Feichtinger, 1996. Expression of vimentin, cytokeratin and desmin in Sertoli cells of human fetal, cryptorchid and tumour-adjacent testicular tissue. Virchow Arch., 427: 497-502.
PubMedDirect Link - Sadri-Ardekani, H., S.C. Mizrak, S.K.M. van Daalen, C.M. Korver and H.L. Roepers-Gajadien et al., 2009. Propagation of human spermatogonial stem cells in vitro. JAMA., 302: 2127-2134.
Direct Link - Scarpino, S., A.R. Morena, C. Petersen, B. Froysa, O. Soder and C. Boitani, 1998. A rapid method of Sertoli cell isolation by DSA lectin, allowing mitotic analyses. Mol. Cell. Endocrinol., 146: 121-127.
CrossRef - Schlatt, S., S.S. Kim and R. Gosden, 2002. Spermatogenesis and steroidogenesis in mouse, hamster and monkey testicular tissue after cryopreservation and heterotopic grafting to castrated hosts. Reproduction, 124: 339-346.
CrossRefDirect Link - Schlatt, S., A. Meinhardt and E. Nieschlag, 1997. Paracrine regulation of cellular interactions in the testis: Factors in search of a function. Eur. J. Endocrinol., 137: 107-117.
CrossRefDirect Link - Schlatt, S., G. Rosiepen, G.F. Weinbauer, C. Rolf, P.F. Brook and E. Nieschlag, 1999. Germ cell transfer into rat, bovine, monkey and human testes. Hum. Reprod., 14: 144-150.
CrossRefDirect Link - Stojkovic, M., M. Lako, P. Stojkovic, R. Stewart and S. Przyborski et al., 2004. Derivation of human embryonic stem cells from day-8 blastocysts recovered after three-step in vitro culture. Stem Cells, 22: 790-797.
PubMedDirect Link - Tadokoro, Y., K. Yomogida, H. Ohta, A. Tohda and Y. Nishimune, 2002. Homeostatic regulation of germinal stem cell proliferation by the GDNF/FSH pathway. Mech. Dev., 113: 29-39.
PubMedDirect Link - Tehranipour, M., 2010. Bone marrow stromal cells for the treatment of spinal cord injury in rats. J. Biol. Sci., 10: 53-57.
CrossRefDirect Link - Van der Wee, K.S., E.W. Johnson, G. Dirami, T.M. Dym and M.C. Hofmann, 2001. Immunomagnetic isolation and long-term culture of mouse type A spermatogonia. J. Androl., 22: 696-704.
Direct Link - Wahab-Wahlgren, A., N. Martinelle, M. Holst, K. Jahnukainen, M. Parvinen and O. Soder, 2003. EGF stimulates rat spermatogonial DNA synthesis in seminiferous tubule segments in vitro. Mol. Cell. Endocrinol., 201: 39-46.
PubMedDirect Link - Ward, J.A., J. Robinson, B.J. Furr, S.M. Shalet, I.D. Morris, 1990. Protection of spermatogenesis in rats from the cytotoxic procarbazine by the depot formulation of Zoladex, a gonadotropin-releasing hormone agonist. Cancer Res., 50: 568-574.
PubMedDirect Link - Watt, F.M. and B.L. Hogan, 2000. Out of Eden: Stem cells and their niches. Sci., 287: 1427-1430.
Direct Link