
Raden Lukas Martindro Satrio Ari Wibowo
Postgraduate School of Animal Science, Faculty of Animal Science, Universitas Gadjah Mada, Jl. Fauna No. 3, Bulaksumur, 55281 Yogyakarta, Indonesia
Zaenal Bachruddin
Department of Animal Nutrition and Feed Science, Faculty of Animal Science, Universitas Gadjah Mada, Jl. Fauna No. 3, Bulaksumur, 55281 Yogyakarta, Indonesia
Nanung Agus Fitriyanto
Department of Animal Products Technology, Faculty of Animal Science, Universitas Gadjah Mada, Jl. Fauna No. 3, Bulaksumur, 55281 Yogyakarta, Indonesia
LiveDNA: 62.12639
Tomoyuki Nakagawa
Departement of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, Gifu, Japan
Takashi Hayakawa
Departement of Applied Life Science, Faculty of Applied Biological Sciences, Gifu University, Gifu, Japan
Ambar Pertiwiningrum
Department of Animal Products Technology, Faculty of Animal Science, Universitas Gadjah Mada, Jl. Fauna No. 3, Bulaksumur, 55281 Yogyakarta, Indonesia
LiveDNA: 62.16078
Pakistan Journal of Nutrition
Year: 2017 | Volume: 16 | Issue: 7 | Page No.: 488-496







ABSTRACT
Background: Puffer fish skin tannery is an alternative to substitute the production of hide and animal skin in Indonesia that has been decreasing. To improve the quality of puffer fish leather, keratinase was needed by removing the thorns. Objective: The aim of this study was to screen bacteria that show keratinolytic activity. Methodology: The isolated bacterial strains were screened for the production of extracellular keratinase using skim milk agar. After incubation, the formations of a clear zone around the bacterial growth were observed. They were identified based on morphological enzyme activity and molecular identification. Bacterial screening and identification were conducted with descriptive method. The microbial activity tests were analyzed using a completely randomized design. Results: The result of the screening was that 3 of 5 kinds of strains exhibited caseinolytic activity (by showing the clear zone). Three Bacillus bacteria that were newly isolated from puffer fish waste-using a feather enrichment technique-were identified on the basis of 16S ribosomal RNA gene sequence analysis, physiological and carbohydrates assimilation tests. They were revealed as the strains of Bacillus thuringiensis for BRAW_PT isolate, Bacillus aerius for BRAW_PB isolate and Bacillus firmus for BRAW_PI isolate. The results of proteolytic enzymes assay showed that Bacillus firmus BRAW_PI has the highest protease and keratinase activity, which was 37.52±0.96 and 6.781±0.479 U mg–1 consecutively. Conclusion: All bacteria obtained were the superior bacteria that can be used for the removal of thorns from puffer fish skin in the tanning process.
PDF Abstract XML References Citation
Received: March 14, 2017;
Accepted: May 18, 2017;
Published: June 15, 2017
Copyright: © 2017. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Raden Lukas Martindro Satrio Ari Wibowo, Zaenal Bachruddin, Nanung Agus Fitriyanto, Tomoyuki Nakagawa, Takashi Hayakawa and Ambar Pertiwiningrum, 2017. Screening and Characterization of Keratinolytic Bacteria from Puffer Fish Skin Waste. Pakistan Journal of Nutrition, 16: 488-496.
DOI: 10.3923/pjn.2017.488.496
URL: https://scialert.net/abstract/?doi=pjn.2017.488.496
DOI: 10.3923/pjn.2017.488.496
URL: https://scialert.net/abstract/?doi=pjn.2017.488.496
INTRODUCTION
Leather and its processing industry is one of the leading industries in Indonesia1. It is reflected in the large contribution of the industry to national fund. Although the leather industry is considered as important field, there are many issues, which have to improve by the government, business people and other stakeholders, including higher education institutions.
One of the issues in leather industry is limited fresh leather material. Indonesia has a shortage of material supply to fulfill domestic demands and then the amount of import of raw- and finished-leather material keeps increasing in order to fulfill domestic demands of them2. The rarity of leather material is experienced by industry players since Decree of The President of The Republic of Indonesia No. 46/1997, which states that raw material can only be imported from countries free from contagious animal diseases included in the A list of Office International des Epizootis. Since the regulation was implemented, an animal product imported to Indonesia has been under strict monitoring to prevent contagion or entry of contagious diseases, such as foot and mouth disease and rinderpest. Government Regulation of The Republic of Indonesia No. 82/2000 Article 30 on Animal Quarantine authorizes rejection if an animal comes from forbidden country or area3. Considering the size of Indonesia’s waters is bigger than the land, which is around 5.8 million km2, fishery production in Indonesia is quite large and so is the number of fish type (commodity) caught4.
Puffer fish (Arothron reticularis) is an underutilized fish in any fishing. It can be superior product for the surrounding communities. One of the utilization of puffer fish skin is for tannery business. The tanning of puffer fish leather can be an alternative to tanning industry that is currently limited to the production of cattle leather. Fish tanning business doesn’t only give value added to leather waste but also become an alternative in fulfilling leather material in the leather industry in Indonesia. Additionally, puffer fish also has a uniqueness that lies in its rounded body shape5.
The quality of leather from puffer fish can be improved by removing its spines. One of the ways to destroy the spines is to degrade the protein keratin in it with the keratinase enzyme. Keratinase is important for the pre-tanning process in leather industry so the skin tanning can be an eco-friendly process by reducing the use of sodium sulfate6.
Keratinase that is produced by microbes is an enzyme capable of degrading the structural protein that is generally found in feathers, hair and wool. The types of bacteria that have been screened to produce extracellular keratinase that can degrade fur and keratin, are for example Bacillus licheniformis, Bacillus subtilis, Bacillus cereus and Bacillus pseudofirmus7. Therefore, this study was performed to screen and to characterize the keratinolytic bacteria as one of the innovative solutions in tannery processing and become an interesting major to be developed and studied in depth.
MATERIALS AND METHODS
Materials: Materials used in this research were 30 pieces of 14-days-rotten puffer fish skin. The skin was taken randomly from the fish market in TPI Tanjungsari of Marine and Fisheries Agency of Rembang. It is used as raw material for the isolation and identification of microbes.
The growth medium for the isolation process was the same as the one used by Macedo et al.8 with modification: Puffer fish skin flour as the sole carbon and nitrogen sources as much as 10 g and as mineral sources are: 0.5 g of NaCl, 0.3 g of K2HPO4 and 0.4 g of KH2PO4. The medium for the stock solution includes 1 g of yeast extract, 1 g of biological peptone, 0.5 g of NaCl and 100 mL of H2O.
The materials for microbial identification are: (a) Simple staining including 70% alcohol, safranin, Gram staining including, gram A (crystal violet), gram B (Lugol iodine), H2O, gram C (acetone alcohol washing solution), gram D (safranin), Ziehl Neelsen’s stain includes ZN A carbol fuchsin solution as the primary stain, ZN B acid alcohol solution (37%) as a laxative and ZN C Loeffler’s methylene blue solution. The materials for molecular identification consist of tris-HCl, NaCl, EDTA, SDS, proteinase K, phenol, chloroform, TE, ethanol, RNAase, PCR extraction kit (Genaid) , sequencing kit, primary ABI PRISM 3730xl Genetic Analyzer develop by Applied Biosystems, USA. Kit that was used for the sequencing purpose is BigDye® Terminator v3.1 .
The equipments used in this research are: HIRAYAMA Vertical Autoclaves autoclave-(Tokyo, Japan), LABCONCO Horizontal laminar air flow (Kansas, USA), MEMMERT oven (Schwabach, Germany), MEMMERT water bath (Schwabach, Germany), SOCOREX macro and micropipettes (Socorex Isba S.A. Switzerland), HANNA pH meter (Michigan, USA), THERMOLYNE stirrer (Atlanta, USA), OHAUS analytical balances (Shanghai, China), MOTIC test tubes, loopful (Wertheim-Mondfel 97877, Germany), microscopes MOTIC Wertheim-Mondfel 97877, SNE 4500M SEM microscope-Pleasanton, CA, PERKIN ELMER Lambda 25 UV/Vis Spectrophotometer (Waltham, MA 02451, USA), ROTOFIX 32 coolbox and centrifuge (California, USA), EPPENDORF 5804R refrigerated centrifuge (California, USA), PCR BIO-RAD T100 Thermal Cycler sequencer Applied Biosystems, (USA), STUART shaker (Staffordshire, UK), Glove Box (China), MEMMERT (Schwabach, Germany) and HERAEUS incubators (UK/Ireland), IWAKI glasswares, (Fukushima, Japan) and HERMA glasswares, (Germany) glasswares, such as Erlenmeyer, petri dishes, glass slide, bunsen, glass beaker, measuring glass and flask. All reagents were of analytical reagent grade.
Screening of keratinolytic bacteria: The isolated bacterial strains were screened for the production of extracellular keratinase using skim milk agar medium. The pure cultures were streaked on the skim milk agar plates and the plates were incubated at 37°C for 48 h. After incubation, the formations of a clear zone around the bacterial growth were observed9.
Caseinolytic activity: Skim milk agar medium was sterilized at 121°C for 15 min at 15 lbs pressure. The isolates were streaked on the medium. The zone formed around the colonies due to the production of caseinase enzyme was considered as a positive result. The organisms screened with skim milk agar medium were subcultured by growing the bacterium in nutrient broth medium at 37°C for 24 h10.
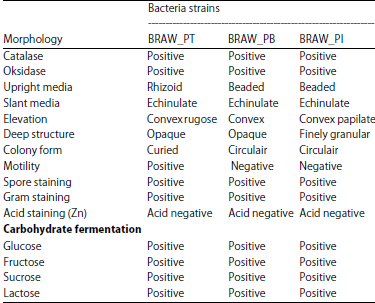
Morphological test: The identification of bacteria can be conducted by viewing the colony morphology both macroscopically and microscopically, including simple, Gram, Zn and spore staining as well as viewing on the bacterial biochemical test. The morphology of bacteria includes the shape, size, texture, colony color and motility. The biochemical test was conducted to ensure the species of bacteria, it included the test of catalase, oxidase and fermentation of carbohydrates.
Production of keratinolytic enzymes: The enzyme production was based on the method of Hoq et al.11 with slight modification. Each of the isolates was cultivated in a basal medium (per liter of the solution: NaCl, 0.5 g: 0.06 g: K2HPO4, 0.3 g: K2HPO4, 0.4 g) containing keratinous substrates: (10.0 g of puffer fish, pH 7.5) as the only source of nitrogen, carbon and sulfur. The supplementation of 1.0 g of nitrogen (yeast extract) and 1.0 g of carbon sources (bacteriological peptone) together was also tested in presence or absence of keratinous substrates under identical conditions. Cultivation was done with 5 mL of 24 h grown inoculum (in nutrient broth) of the respective bacterial cultures (100 mL) on the liquid medium in a 500 mL Erlenmeyer flask at 37°C under shaking (120 rpm) for 24 h. The samples were selected after 24 h and centrifuged at 4500 rpm at 6°C for 20 min. The supernatants were preserved at 4°C and assayed for protein and enzymes.
One ose pure isolates was cultured in 5 mL of sterile Pre-culture medium (Stock solution) and incubated at 30°C for 48 h in a shaker incubator at 120 rpm. After incubation, the broth was centrifuged at 4500 rpm for 20 min and the supernatant was used to study the keratinolytic activities.
Protease activity: Protease activity was determined with the method of Bergmeyer et al.12 One unit (U) of proteolytic enzyme activity is defined as the amount of enzyme that releases 1 μmol tyrosine per milliliter per minute at standard test condition. Specific activity is stated in enzyme activity unit per milligram of protein.
Keratinolytic activity: Keratinolytic activity was determined with the method of Wang et al.13. The substrate used was keratin azure. The keratinase enzyme activity was measured by using several types of keratin substrates such as feather flour,14-16 and keratin azure.17,18,13 Keratin azure is pure keratin derived from wool that is stained with azo dyes. Keratin azure that is added with water and is reacted with specific enzyme will produce a blue-colored reaction product. The amount of keratinase enzyme activity is determined by Unit, where one unit (U) of keratinase activity is defined as the amount of enzyme required to improve the absorbance of 0.01 between the sample and the control at a wavelength of 595 nm in accordance with the test conditions.19,13
Scanning Electron Microscope (SEM): The fixation solution was made by using 0.2145 g of sodium cacodylic, 1.0081 g of NaCl, 630 μL of HCl 0.2 M, 1 mL of glutaraldehyde and was added with H2O for a volume of 100 mL using a Volumetric flask. After all of the ingredients were dissolved and mixed together until the mixture was homogenous, the solution was put into a dark bottle and stored in the refrigerator.
Agar medium was made using 3.5 g of agar, 0.1 g of gelatin and added with 1/100 stock solution for dilution that consisted of 1 mL of stock solution added with 99 mL of H2O. The 100% stock solution was made by putting 1 g of meat extract, 1 g of microbiological peptone, 0.5 g of NaCl and 80 mL of H2O into a glass beaker. It was then stirred well until it dissolved evenly and the pH was adjusted to 7.2. If it is too acidic, it can be added with 0.1 N of NaOH and if it is too alkaline, it can be added with 0.1 N of H2SO4. Then, the solution was poured into the Erlenmeyer and added with H2O to get 100 mL volume. It was then boiled on the stove and stirred constantly until it dissolved evenly. After that, it was sterilized with an autoclave.
The sterilized agar medium was then poured into the petri dish. A special filter paper that had been cut to the size of 5×5 mm (previously sterilized) was put on the un-ossified agar and we waited until it was cool and ossified. The isolates which had previously been grown on preculture medium and shaken for one night was taken 1 μL and dripped on the filter paper that had been placed on the agar, then it was incubated for 4 days.
Filter paper that had been overgrown by isolates on the agar was then taken and put in microcentrifuge tube, added with 1 mL of fixation solution and incubated at 4°C for 30 min. The fixation solution was then taken using a pipette until it was empty, added with 1 mL of acetone 60% and incubated at room temperature for 15 min. The acetone 60% was then taken until it was empty and replaced by 1 mL of acetone 80%. The acetone 80% was then taken until it was empty and replaced by 1 mL of acetone 100%, the process with acetone 100% was repeated twice. After the acetone had been removed and replaced by 1 mL of pentyl acetate and stored at room temperature for 20 min, it was dried using filter vacuum. When the fluid was gone, the sample had reached the critical point drying. It was then coated and ready to be observed using a SEM20.
Molecular identification: The molecular identification was conducted using the 16S rRNA gene sequence. The sequencing of 16S-rRNA gene consisted of several stages, including DNA extraction, amplification between 16S-rRNA gene and PCR, then sequencing using ABI PRISM 3730xl Genetic Analyzer developed by Applied Biosystems, USA. Kit that we use for the sequencing purpose is BigDye® Terminator v3.1 .
Amplification of 16S rRNA gene was conducted using thermal cycler. The Primers used were the couple of 8F (5’-AGAGGTTGATCCTGGCTCAG-3’), primer 1492R (5’-GTTTACCTTGTTACGACTT-3’). The PCR process began with initial denaturation stage at a temperature of 94°C for 5 min and continued with the process of as many as 30 cycles consisted of denaturation process at a temperature of 94°C for 1 sec, primer attachment at a temperature of 55°C for 1 min and elongation at 72°C for 1 min. After the 30 cycles had finished, it was followed by lengthening process on the temperature of 72°C for 10 min and the PCR process stopped at a temperature of 12°C. PCR results were then viewed by electrophoresis on agarose gel of 0.8%.
Phylogenetic analysis: The identical 16S rDNA sequences were identified by phylogenetic tree analysis and manual comparison, where the sequences with a similarity of more than 90% were described as identical and these sequences were used for further phylogenetic analysis as Operational Taxonomic Unit (UTO). The evolutionary tree was based on distances compiled using the nearest neighbor algorithm.
Data analysis: Bacterial isolation and identification were conducted with descriptive method. The data of microbial activity obtained were described as the Mean±SD. Data were analyzed using one-way analysis of variance (ANOVA) followed by Duncan’s Multiple-Range (DMR) test using software SPSS Inc., (Chicago, IL, USA). Differences considered significant when the probability was less than 5%.
RESULTS
Totally five isolates were found from puffer fish waste. All the isolates were subjected to primary screening on Milk Agar plate and 3 of 5 isolates formed the clear zone, which supported the degradation and utilization of casein (Skim Milk) by the respective isolates. Those organisms were named as BRAW–PT, BRAW–PB and BRAW–PI strains (BRAW-Buntal Rembang Ari Wibowo). The macroscopic observation on bacterial isolates is shown in Table 1.
They capable of growing and degrading puffer fish skin at 35°C within 14 days. The BRAW–PT, BRAW–PB and BRAW–PI strains which appeared single or in the chain had straight rods. They were Gram-positive, endospore-forming organisms, aerobic, motile, strong oxidase and catalase positive. The additional morphological, physiological and biochemical test are shown in Table 2.
Caseinolytic activity: Table 3 shows the results of clear zone diameter in 72 h of observations, BRAW–PT strains showed the largest colony diameter (3.06 mm), followed by BRAW–PI strain (2.40 mm). The smallest colony diameter (1.34 mm) belonged to BRAW–PB strain. The results of clear zone diameter and the diameters of colonies from the largest to the smallest were then sorted and tested to determine the protease activity.
Proteolytic activity: The results of proteolytic enzyme assay showed that BRAW–PI had a specific activity of 37.52±0.96 U mg–1 (Table 4).
| Table 1: | Observation of keratinase bacterial isolates |
 | |
| Table 2: | Morphology of keratinolytic isolates |
 | |
| Table 3: | Diameter of clear zone |
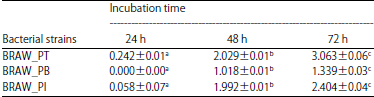 | |
| a-cDifferent superscripts in the same column indicate significant differences (p<0.05) | |
| Table 4: | Proteinase enzyme activity |
 | |
a,bDifferent superscripts in the same column indicate significant differences (p<0.05) | |
| Table 5: | Keratinase enzyme activity |
 | |
a,bDifferent superscripts in the same column indicate significant differences (p<0.05) | |
This was the highest activity of the three isolates. The test showed significantly different results among the treatments. A further test using Duncan’s Multiple Range Test (DMRT) showed that BRAW–PI had a significantly different proteolytic enzyme activity (p<0.05) than other isolates. Proteinase enzyme activity of BRAW–PT and BRAW–PB had no significant differences (p<0.05).
Keratinase activity: Keratinase activity assay was carried out on all of 3 isolates. The test results of keratinase activity are shown in Table 5. Based on the Table 5, BRAW–PI strain had the highest (6.78 U mg–1) specific keratinase activity.
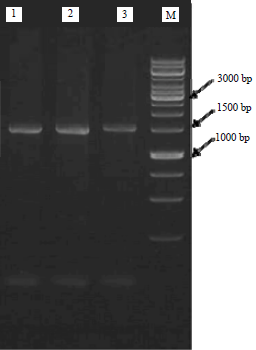 | |
| Fig. 1: | Agarose gel electrophoresis of amplified products |
| Lane 1: BRAW–PI, Lane 2: BRAW–PB, Lane 3: BRAW–PT, M: Broad range marker | |
The BRAW–PI had a significantly different proteolytic enzyme activity (p<0.05) than other isolates. The proteinase enzyme activity of BRAW–PT and BRAW–PB had no significant differences (p<0.05).
Molecular taxonomy, sequencing and phylogenetic analysis: Results of PCR with 16S rRNA primer was then viewed by electrophoresis on a 0.8% agarose gel as shown in Fig. 1. The isolates had a very close genetic relationship with Bacillaceae family based on the 16S rRNA gene sequencing method. BRAW–PT was closed to Bacillus thuringiensis by showing 99% similarity, BRAW–PB was closed to Bacillus aerius by showing 99% similarity and BRAW–PI was closed to Bacillus firmus by showing 99% similarity. The phylogenetic tree (Fig. 2) was constructed by the neighbor-joining method using Molecular Evolutionary Genetics Analysis 6 (MEGA6) program.
DISCUSSION
Skim milk contains casein, a milk protein which will be degraded by proteolytic microorganisms into dissolved nitrogen compounds so the colony will be surrounded by a clear area. It showed that these microbes had proteolytic activity21. Based on this test, there were only three isolates that had the ability to degrade casein isolates while the other two did not.
 | |
| Fig. 2: | Phylogenetic tree of three isolates |
Three of five isolates that showed protease activity were later corroborated by the skim milk agar and they were used for further research. Sivakumar et al.22 confirmed that the zone formed around colonies was due to the formation of the casein enzyme. It was considered as a positive result. The caseinolytic ability of bacteria could be used to select the initial keratinolytic bacteria because most keratinolytic bacteria that derived from nature also had a good caseinolytic activity. The results obtained showed that the BRAW–PB, BRAW–PT and BRAW–PI isolates were able to degrade the casein because casein is the main protein in milk. Benson23 stated that the media became clear due to the caseinase exoenzymes produced by bacteria.
Based on the method of Gupta and Ramnani9, the chosen casein agar media was related to the most reported keratinase enzyme derived from nature. The isolates were grown in an incubator at 30°C for 72 h. The statistical analysis showed that there were significant differences among the treatments. Duncan’s Multiple Range Test (DMRT) showed that the clear zone diameter of all isolates had been significantly affected by the incubation time. All isolates had the largest clear zone diameter in 72 h. It can be seen in Table 3.
Brandelli et al.24 stated that the hydrolysis ability of casein depended on the species and environment of the bacterial isolation place. Furthermore, in order to ensure the ability of bacteria in hydrolyzing protein, a protease activity test was conducted. The screening was conducted on 3 isolates using qualitative (the formation of the clear zone) and quantitative (enzymes activity) methods25.
Protease is also called peptidase or proteinase. It is a hydrolase-class enzyme that will break down proteins into simpler molecules such as short oligopeptides or amino acids, with hydrolysis reaction on the peptide bond. Proteolytic bacteria are the bacteria that are able to produce extracellular protease enzyme. The enzyme breaks protein that is produced in the cell and releases it out of the cell.
Some researcher found keratinase activity in some bacterial enzymes such as Bacillus subtilis that has keratinase activity26 of 1.8 U mL–1, Aspergillus sp. 1.7 U mL–1, Cladosporium sp. 1.9 U mL–1, Periconia sp.27 1.5 U mL–1 as well as Streptomyces gulbargensis that has keratinase activity18 of 1.5 U mL–1. Nevertheless, the keratinase activity of those three strains is lower than the one produced by Bacillus megaterium28 F7-1 by 58 U mL–1, Chryseobacterium sp.29 by 40 U mL–1 and Flavobacterium sp.30 by 7 U mL–1.
Some keratinolytic microorganisms have been reported, including several species of fungi such as Microsporum31, Trichophyton32, Streptomyces33,34 and Actinomycetes35,36. Recently, keratinase activity was also reported for coccus that was rod-shaped Gram-positive.
Yamamura et al.37 reported a joint action between the similar protein disulfide reductase and proteases produced by Stenotrophomonas sp. to degrade deer-hair keratin. The mechanisms of keratin degradation occurred as follows:
![]()
There were two types of morphological forms of microbes in the form of short bacilli, while four types of microbes are long-shaped bacilli. The SEM was useful to clarify the simple coloring using safranin dye and Gram and Zn staining indicated that all of the isolates belonged to Bacilli class.
The results of amplification of the encoding 16S rRNA gene of bacterial isolate were then determined for its DNA base sequences. The process of determining the base sequence of DNA was conducted by 1st BASE, Malaysia. The results of the determination was then read with DNA baser. Then, the DNA’s base sequence obtained was used to search for the comparison of DNA sequence in various similar microorganisms or those that have a close genetic relationship to the NCBI (National Center for Biotechnology Information) GenBank through the BLAST (Basic Local Alignment Search Tool) method (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The results of the matching using BLAST method was then selected to find the one that had the closest genetic relationship and sequenced for the phylogenetic tree using the Molecular Evolutionary Genetics Analysis 6 (MEGA6) program. The sequence of 16S rRNA gene has been determined for many strains. Genbank is the largest data bank for nucleotide sequences, saving over 20 million nucleotide sequences and almost more than 90.000 of them are the 16S rRNA gene. It shows that many previously saved nucleotide sequences are compared with the sequence of a newly known strain. In addition, the universal 16S rRNA gene in bacteria can be used to analyze the phylogenetic relationship between the bacteria from the genus level of many phyla to the level of strains that are species and subspecies.
The genetic relationship of bacteria was known from the base sequence analysis of 16S rRNA gene nitrogen. The base sequences of the isolates’ nitrogen and the base sequences of the reference strain nitrogen or comparators were used to analyze the genetic relationship in the form of a phylogenetic tree. The bootstrap value was indicated by the number contained in the branches of the phylogenetic tree. According to Hall38, the higher value bootstraps, the more reliability or trustworthiness. The results of phylogenetic tree analysis are shown in Fig. 2.
Based on the formed phylogenetic tree, all isolates had a very close genetic relationship with different species of Bacillaceae family. BRAW–PT was close to Bacillus thuringiensis with 99% similarity, BRAW–PB was close to Bacillus aerius with 99% similarity and BRAW–PI was close to Bacillus firmus with 99% similarity.
CONCLUSION
The results of the screening showed that 3 of 5 bacterial strains presented keratinolytic activity. They are Bacillus thuringiensis BRAW–PT, Bacillus aerius BRAW–PB and Bacillus firmus BRAW–PI. The proteolytic enzyme test showed that BRAW–PI bacterial strain had the highest protease and keratinolytic specific activity, which was 37.52±0.96 and 6.781±0.479 U mg–1 consecutively.
SIGNIFICANCE STATEMENTS
The study managed to discover bacteria with the ability to degrade keratin of pufferfish skin, which is helpful for an environmentally friendly tanning process. The bacteria that discovered in this study was the result of independent isolation. The innovation of bacteria with keratinolytic activities from decaying pufferfish’s skin has never been conducted before. It also could use to process on pufferfish skin tanning which also never been conducted before. This study will help the researcher to overcome environmental pollution resulting from tanning processes which have not been widely studied by other researchers. Thus, this innovation i.e. enzymes produced by bacteria which cause pufferfish’s skin to decay, can be used as a substitute for chemicals in tanning processes.
REFERENCES
- Mohamed, K.S., T.V. Sathianandan, V. Kripa and P.U. Zacharia, 2013. Puffer fish menace in Kerala: A case of decline in predatory control in the Southeastern Arabian sea. Curr. Sci., 104: 426-429.
Direct Link - Thanikaivelan, P., J.R. Rao, B.U. Nair and T. Ramasami, 2005. Recent trends in leather making: Processes, problems and pathways. Crit. Rev. Environ. Sci. Technol., 35: 37-79.
CrossRefDirect Link - Pillai, P., S. Mandge and G. Archana, 2011. Statistical optimization of production and tannery applications of a keratinolytic serine protease from Bacillus subtilis P13. Process Biochem., 46: 1110-1117.
CrossRefDirect Link - Macedo, A.J., W.O.B. da Silva, R. Gava, D. Driemeier, J.A.P. Henriques and C. Termignoni, 2005. Novel keratinase from Bacillus subtilis S14 exhibiting remarkable dehairing capabilities. Applied Environ. Microbiol., 71: 594-596.
CrossRefDirect Link - Gupta, R. and P. Ramnani, 2006. Microbial keratinases and their prospective applications: An overview. Applied Microbiol. Biotechnol., 70: 21-33.
CrossRefDirect Link - Hoq, M.M., K.A.Z. Siddiquee, H. Kawasaki and T. Seki, 2005. Keratinolytic activity of some newly isolated Bacillus species. J. Biol. Sci., 5: 193-200.
CrossRefDirect Link - Wang, S.L., W.T. Hsu, T.W Liang, Y.H Yen and C.L Wang, 2008. Purification and characterization of three novel keratinolytic metalloproteases produced by Chryseobacterium indologenes TKU014 in a shrimp shell powder medium. Bioresour. Technol., 99: 5679-5686.
CrossRefDirect Link - Rahayu, S., D. Syah and M.T. Suhartono, 2012. Degradation of keratin by keratinase and disulfide reductase from Bacillus sp. MTS of Indonesian origin. Biocatal. Agric. Biotechnol., 1: 152-158.
CrossRefDirect Link - Cedrola, S.M.L., A.C.N. de Melo, A.M. Mazotto, U. Lins and R.B. Zingali et al., 2012. Keratinases and sulfide from Bacillus subtilis SLC to recycle feather waste. World J. Microbiol. Biotechnol., 28: 1259-1269.
CrossRefDirect Link - Mazotto, A.M., A.C.N. de Melo, A. Macrae, A.S. Rosado and R. Peixoto et al., 2011. Biodegradation of feather waste by extracellular keratinases and gelatinases from Bacillus spp. World J. Microbiol. Biotechnol., 27: 1355-1365.
CrossRefDirect Link - Jaouadi, B., B. Abdelmalek, D. Fodil, F.Z. Ferradji, H. Rekik, N. Zarai and S. Bejar, 2010. Purification and characterization of a thermostable keratinolytic serine alkaline proteinase from Streptomyces sp. strain AB1 with high stability in organic solvents. Bioresour. Technol., 101: 8361-8369.
CrossRefDirect Link - Syed, D.G., J.C. Lee, W.J. Li, C.J. Kim and D. Agasar, 2009. Production, characterization and application of keratinase from Streptomyces gulbargensis. Bioresour. Technol., 100: 1868-1871.
CrossRefDirect Link - Cai, C.G., J.S. Chen, J.J. Qi, Y. Yin and X.D. Zheng, 2008. Purification and characterization of keratinase from a new Bacillus subtilis strain. J. Zhejiang Univ. Sci. B, 9: 713-720.
CrossRefDirect Link - Sivakumar, T., T. Shankar, P. Vijayabaskar and V. Ramasubramanian, 2012. Optimization for keratinase enzyme production using Bacillus thuringiensis TS2. Acad. J. Plant Sci., 5: 102-109.
Direct Link - Brandelli, A., D.J. Daroit and A. Riffel, 2009. Biochemical features of microbial keratinases and their production and applications. Applied Microbiol. Biotechnol., 85: 1735-1750.
CrossRefPubMedDirect Link - Pillai, P. and G. Archana, 2008. Hide depilation and feather disintegration studies with keratinolytic serine protease from a novel Bacillus subtilis isolate. Applied Microbiol. Biotechnol., 78: 643-650.
CrossRefPubMedDirect Link - Eliades, L., M. Cabello, C. Voget, B. Galarza and M. Saparrat, 2010. Screening for alkaline keratinolytic activity in fungi isolated from soils of the biosphere reserve Parque Costero del Sur (Argentina). World J. Microbiol. Biotechnol., 26: 2105-2111.
CrossRefDirect Link - Park, G.T. and H.J. Son, 2009. Keratinolytic activity of Bacillus megaterium F7-1, a feather-degrading mesophilic bacterium. Microbiol. Res., 164: 478-485.
CrossRefDirect Link - Riffel, A., F. Lucas, P. Heeb and A. Brandelli, 2003. Characterization of a new keratinolytic bacterium that completely degrades native feather keratin. Arch. Microbiol., 179: 258-265.
CrossRefDirect Link - Riffel, A. and A. Brandelli, 2006. Keratinolytic bacteria isolated from feather waste. Braz. J. Microbiol., 37: 395-399.
CrossRefDirect Link - Essien, J., A. Umoh, E. Akpan, S. Eduok and A. Umoiyoho, 2009. Growth, keratinolytic proteinase activity and thermotolerance of dermatophytes associated with alopecia in Uyo, Nigeria. Acta Microbiol. Immunol. Hung., 56: 61-69.
CrossRefDirect Link - Anbu, P., S.C.B. Gopinath, A. Hilda, N. Mathivanan and G. Annadurai, 2006. Secretion of keratinolytic enzymes and keratinolysis by Scopulariopsis brevicaulis and Trichophyton mentagrophytes: Regression analysis. Can. J. Microbiol., 52: 1060-1069.
CrossRefPubMedDirect Link - Szabo, I., A. Benedek, M.I. Szabo and G. Barabas, 2000. Feather degradation with a thermotolerant Streptomyces graminofaciens strain. World J. Microbiol. Biotechnol., 16: 253-255.
CrossRefDirect Link - Tatineni, R., K.K. Doddapanem, R.C. Potumarthi, R.N. Vellanki, M.T. Kandathil, N. Kolli and L.N. Mangamoori, 2008. Purification and characterization of an alkaline keratinase from Streptomyces sp. Bioresour. Technol., 99: 1596-1602.
CrossRefPubMedDirect Link - Young, R.A. and R.E. Smith, 1975. Degradation of feather keratin by culture filtrates of Streptomyces fradiae. Can. J. Microbiol., 21: 583-586.
CrossRefPubMedDirect Link - Bockle, B., B. Galunsky and R. Muller, 1995. Characterization of a keratinolytic serine proteinase from Streptomyces pactum DSM 40530. Applied Environ. Microbiol., 61: 3705-3710.
PubMedDirect Link - Yamamura, S., Y. Morita, Q. Hasan, K. Yokoyama, E. Tamiya, 2002. Keratin degradation: a cooperative action of two enzymes from Stenotrophomonas sp. Biochem. Biophys. Res. Commun., 294: 1138-1143.
CrossRefDirect Link - Hall, B.G., 2013. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol., 30: 1229-1235.
CrossRefDirect Link