
M.D. Fakruddin
Industrial Microbiology Laboratory, Institute of Food Science and Technology, Bangladesh Council of Scientific and Industrial Research, Dhaka, Bangladesh
Mahmuda Sultana
Biotechnology Program, Department of Mathematics and Natural Sciences, BRAC University, Dhaka, Bangladesh
Monzur Morshed Ahmed
Industrial Microbiology Laboratory, Institute of Food Science and Technology, Bangladesh Council of Scientific and Industrial Research, Dhaka, Bangladesh
Abhijit Chowdhury
Industrial Microbiology Laboratory, Institute of Food Science and Technology, Bangladesh Council of Scientific and Industrial Research, Dhaka, Bangladesh
Naiyyum Choudhury
Biotechnology Program, Department of Mathematics and Natural Sciences, BRAC University, Dhaka, Bangladesh
Pakistan Journal of Biological Sciences
Year: 2013 | Volume: 16 | Issue: 6 | Page No.: 267-274







ABSTRACT
The coastal aquaculture mainly shrimps constitute major export sector in Bangladesh and is increasingly shaped by international trade conditions and by national responses to those stringent quality and safety standards. PCR based validated methods for detection of major bacterial pathogens in shrimp might be very useful tool for ensuring quality and safety standards of exportable shrimps. The objective of this study was to evaluate overall performance (sensitivity and specificity) of the multiplex PCR assay for detection of Vibrio cholerae, Vibrio parahaemolyticus, Salmonella sp. and Escherichia coli O157:H7 from spiked shrimp samples. The targeted genes were ompW for V. cholerae, tdh for V. parahaemolyticus, sefA for Salmonella spp. and hlyEHEC for E. coli O157:H7. The genomic DNA was extracted by using standard method and amplified accordingly. Sensitivity of the assay was tested by inoculating the shrimp homogenate with viable cells of laboratory references strains (target pathogens). The genes were amplified individually both from culture homogenate and spiked samples. Twenty different uniplex and multiplex PCR assay were performed; the results showed that the sensitivity and specificity of multiplex PCR are comparable to that of the results of uniplex PCR for the samples. DNA extracted from shrimp samples spiked with non-target pathogen (Bacillus cereus, Shigella flexneri and Staphylococcus aureus) yielded negative results.
PDF Abstract XML References Citation
Received: December 04, 2012;
Accepted: February 14, 2013;
Published: March 21, 2013
How to cite this article
M.D. Fakruddin, Mahmuda Sultana, Monzur Morshed Ahmed, Abhijit Chowdhury and Naiyyum Choudhury, 2013. Multiplex PCR (Polymerase Chain Reaction) Assay for Detection of E. coli O157:H7, Salmonella sp., Vibrio cholerae and Vibrio parahaemolyticus in Spiked Shrimps (Penaeus monodon). Pakistan Journal of Biological Sciences, 16: 267-274.
DOI: 10.3923/pjbs.2013.267.274
URL: https://scialert.net/abstract/?doi=pjbs.2013.267.274
DOI: 10.3923/pjbs.2013.267.274
URL: https://scialert.net/abstract/?doi=pjbs.2013.267.274
INTRODUCTION
Shrimp is a very popular and common food item worldwide especially in western countries and is an important export comodity for many countries like Bangladesh. Presently Bangladesh contributes around 5% to the total global cultured shrimp production. There are nearly 1 million people employed in the shrimp sector in Bangladesh and generating over US$1.2 billion annually (Yunus, 2009). Shrimp is usually consumed uncooked and/or minimally processed, which makes it potential health risk food (Ahmed and Anwar, 2007). Hence, quality of shrimp is a major concern for both consumers and exporters as microbial pathogens from shrimp may be transmitted to humans when they are eaten undercooked or minimally processed or when they transfer the pathogen to other food products through cross-contamination (Khan et al., 2007). According to the guideline of ICMSF (International Commission on Microbiological Specification for Foods) and EU Standard, shrimp samples must be free of pathogens like E. coli O157:H7, Vibrio cholerae, Vibrio parahaemolyticus, Salmonella sp., Coagulase-Positive Staphylococcus aureus etc. (Ahmed and Anwar, 2007). The availability of reliable detection methods is lengthy and critical for identifying these pathogenic bacteria in shrimp. Selective media based methods for detection and identification of E. coli O157:H7, Vibrio cholerae, Vibrio parahaemolyticus, Salmonella sp. in shrimp are generally time-consuming and thus creates limitation for quick inspection and certification. Furthermore, the pathogens are often present in very low numbers among a background of indigenous microflora and stressed deep frozen matrix, thus rendering recovery of target organisms difficult (Jofre et al., 2005).
Development of PCR based modified methods have been published for rapid, sensitive detection of food-borne pathogens in many previous studies (Hoorfar and Cook, 2003; Rijpens and Herman, 2002; Ahari et al., 2009; AlHaj et al., 2007; Malkawi and Gharaibeh, 2004). Since PCR can target unique genetic sequences such as virulence genes of microorganisms, it also has the potential advantage of being an extremely specific assay (Malorny et al., 2003). On the other hand, conventional methods generally require the use of media containing selective agents for pathogen detection and isolation, it may be difficult to detect injured cells that have the potential to recover and grow when the food is stored/preserved and consumed (Kawasaki et al., 2009). The use of DNA-based techniques enables the detection of pathogenic microbes exposed to stress conditions with greater sensitivity and reliability than conventional culture methods (Mandal et al., 2011). Thus, a sensitive, specific and rapid method that would allow detection of multiple pathogens simultaneously from foods will be useful for food processors and regulatory bodies (Bai et al., 2010).
PCR is the method of choice for detection of microorganisms in food (Sherfi et al., 2006; Seidavi et al., 2008). Simplicity, rapidity, reliability, reproducibility, sensitivity and specificity of PCR make it most competitive (Cheng et al., 2012). Uniplex PCR method amplify single gene at a time while multiplex PCR method simultaneously amplify more than one target sequence (Seidavi et al., 2008). Multiplex PCR method saved considerable time and effort in detection of pathogens. It is now proved that multiplex PCR method have compatible or even superior sensitivity over conventional cultural detection methods (Kawasaki et al., 2005; Yasmin et al., 2007).
The current study is aimed to develop a multiplex PCR method for qualitative detection of Salmonella sp., Vibrio parahaemolyticus, Vibrio cholerae and E. coli O157:H7 directly from spiked shrimp samples and to evaluate the sensitivity of the multiplex PCR method. Based on the results obtained further study will be designed for enhancement of sensitivity, specificity and validation of the method.
MATERIALS AND METHODS
Shrimp samples (Penaeus monodon): Shrimp samples were collected from different shrimp farms located on south-western region of Bangladesh. Shrimps were collected from grow out ponds, brought to the laboratory in iced boxes and stored at -20°C till analysis.
Reference strains and cultures: The bacterial strains used in this experiment were obtained from culture collection pool of Industrial Microbiology Laboratory, IFST, BCSIR. Four reference strains were used for inoculation in shrimp homogenate as well as for spiking of shrimp samples; these are namely Salmonella typhi ATCC-65154, Vibrio parahaemolyticus ATCC-17802, Vibrio cholerae ATCC-15748 and E. coli O157:H7ATCC-12079. Bacterial strains were cultured in Trypticase Soy Broth (TSB,Oxoid) containing 0.6% yeast extract in a shaking incubator at 37°C overnight. The strains were maintained in Trypticase Soy Agar (TSA, Oxoid) slants at 4°C.
Chemicals: PCR reagents were purchased from Promega (Madison, WI, USA). PCR primers were synthesized by 1st Base (Singapore). Bacteriological media and broths were purchased from Oxoid (Hampshire, England). The rest of the materials and chemicals and reagents used in this study were purchased from Sigma Chemical Co. (St Louis, MO, USA).
Selection of genes and primers: Genes selected for this study were tdh for Vibrio parahaemolyticus, ompW for Vibrio cholerae, hlyEHEC for E. coli O157:H7 and sefA for Salmonella sp. Primer sequence and product size of the genes are given in Table 1.
DNA extraction from target organisms: DNA from target organisms was extracted by phenol/chloroform and ethanol precipitation method (Wilson, 1987). Bacterial cells were grown overnight in nutrient broth at 37°C, aerated by shaking at 120 rpm in a shaking incubator. Harvesting was then performed by centrifuging the culture at 10000 rpm for 5 minutes. The supernatant was then discarded and remaining cell pellet was subjected to treatment with DNA extraction solution I (Tris HCl+EDTA+sucrose) for 30 minutes at 37°C on a water bath in order to disrupt cells. Then de-proteinization was done using DNA extraction solution II (proteinase K+SDS+NaCl) at 55°C for 1 h on a water bath. Phenol:chloroform:isoamylalcohol (25:24:1) solvent was used to precipitate proteins. The cell extract was mixed gently with the solvent and the nucleic acids were separated in the aqueous layer by centrifugation at 10000 rpm for 5 min. The aqueous solution of DNA was then removed using micropipette. The DNA was then concentrated by ethanol precipitation in the presence of Sodium acetate. After centrifuging and washing with 70% ethanol solution the final pellet was taken and suspended in TE buffer. This suspension was then stored at 4°C for further use (Fakruddin et al., 2012).
| Table 1: | Primer sequences and product size of the genes targeted |
 | |
DNA extraction from spiked shrimp samples: Before spiking shrimps were peeled and autoclaved, the samples were then spiked with overnight grown bacterial culture (106 CFU mL-1). Following incubation at 37°C for 24 h, spiked shrimps were homogenized with sterile ringer solution in a stomacher. Three milliliter of stomached samples were inoculated into nutrient broth and incubated for 24 h. The overnight grown culture was subjected to DNA extraction and PCR. For extraction of DNA from food samples, method described by Yasmin et al. (2007) was followed.
Quantification and purity of DNA: Quantification of genomic DNA was done using 1.0% agarose gel electrophoresis in 1X TAE buffer followed by staining with ethidium bromide. The concentration of extracted DNA was also estimated by visual comparison of the band with 100 bp marker DNA. The purity and concentration of the extracted DNA was checked by measuring absorbances on T60 UV-VIS spectrophotometer at 260 and 280 nm. Purity was analyzed by absorbance ratio of 260/280 nm (Sahasraundhe and Deodhar, 2010).
Uniplex PCR amplification of tdh, ompW, hlyEHEC and sefA gene: PCR amplification was performed in a 30 μL reaction volume containing 50 ng of DNA template, 3 μL 10X PCR reaction buffer without MgCl2, 0.5 μL 20 mMMgCl2 , 1 μL of dNTP mixture, 1 μL each of forward and reverse primer and 1 unit of Taq polymerase. Same reaction mixture was used for all of the genes. Thermal cycling was done in a DNA engine, BIO-RAD (USA). PCR reactions were maintained for initial denaturation at 94°C for 3 min, followed by 30 cycles of 1 min at 94°C (denaturation), 1 min at 55°C for tdh and ompw, 58°C for sefA and hlyEHEC (annealing) and 1 min at 72°C (extension). The final extension for 9 min at 72°C and hold temperature was maintained at 4°C. PCR products were mixed with 3 μL of 10X loading dye (0.25% bromophenol blue, 0.25% xylene cyanol and 40% sucrose, w/v) and electrophoresis was carried on 1.5% agarose gel in 1X TAE buffer at 100v for 1.5 hr and stained with ethidium bromide (10 μg mL-1). A 50/100 bp DNA ladder was used as a standard molecular weight marker. Reference band sizes for Salmonella, V. parahaemolyticus, V. cholerae and E. coli O157:H7 were 470, 250, 588 and 889 bp, respectively.
Multiplex PCR amplification of tdh, ompW, hlyEHEC and sefA gene: Multiplex PCR was performed in a total volume of 30 μL containing 2 μL of mixed template DNA and 28 μL of PCR master mix composed of 1x PCR buffer, 5.0 mMMgCl2, 25 μM of each of Salmonella sp. detection primers (A058 and A01), 25 μM concentration of each of V. parahaemolyticus detection primers (tdh D3 and tdh D5), 25 μM concentration of each of V. cholerae detection primers (ompW F and ompW R), 25 μM concentration of each of E. coli O157:H7 detection primers (HlyF and HlyR), 200 μMdATP, dCTP, dGTP and dUTP, 0.5 U of Ampli Taq Gold DNA polymerase (Invitrogen, USA) with a DNA thermal cycler. The thermocycler was programmed as 50°C for 2 min for carryover treatment and initial denaturation at 95°C for 5 min. The samples were then subjected to 30 cycles of 95°C for 1 min, 58°C for 1 min, 55°C for 1 min, 54°C for 1 min, 72°C for 1 min and then 72°C for 9 min. The amplified products were then analyzed by 2.0% agarose gel electrophoresis. Reference band sizes for Salmonella, V. parahaemolyticus, V. cholerae and E. coli O157:H7 were 470, 250, 588 and 889 bp, respectively. Two different multiplex set up have been standardized, one for targeting sefA and tdh genes and another for all four genes.
Specificity of the multiplex PCR setting: To investigate the specificity of the multiplex PCR method and to observe that the method do not amplify closely related genes present in other related organisms, Bacillus cereus-ATCC 10876, Shigella flexneri ATCC-12022 and Staphylococcus aureus ATCC-25923 were included in the study. DNA extracted from these organisms was subjected to PCR according to the multiplex settings.
RESULTS
Uniplex amplification and electrophoresis of PCR products: The PCR product of shrimp samples spiked with Vibrio parahaemolyticus ATCC-17802 gave a clear distinct band of 251 bp in size which confirms tdh gene of inoculated V. parahaemolyticus ATCC-17802 (Fig. 1). Samples spiked with Salmonella sp. (SefA gene) showed a distinct PCR product band of 470 bp (Fig. 2) and gave confirmation of sefA gene in the inoculated Salmonella sp.
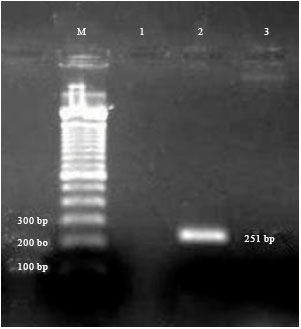 | |
| Fig. 1: | Agarose gel electrophoretic analysis of PCR product of tdh gene of V. parahemolyticus (Lane M: 100 bp DNA ladder , Lane 1: Negative control, Lane 2: tdh gene product (251bp)) |
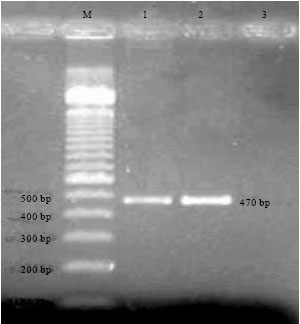 | |
| Fig. 2: | Agarose Gel electrophoretic analysis of PCR product of sefA gene of Salmonella spp. (Lane M: 100 bp DNA ladder, Lane 1 and 2: sefA gene product (470 bp) , Lane 3: Negative control) |
Agarose gel electrophoresis results for both spiked samples with V. cholerae and E. coli O157:H7 showed distinct PCR product band of 588 and 889 bp, respectively (Fig. 3, 4).
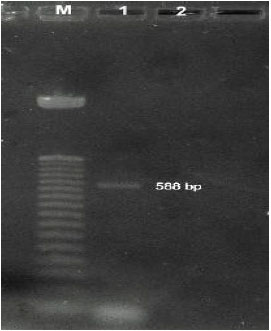 | |
| Fig. 3: | Agarose Gel electrophoretic analysis of PCR product of ompW gene of V. cholerae. (Lane M: 50 bp DNA ladder, Lane 1: ompW gene product (588 bp), Lane 2: Negative control) |
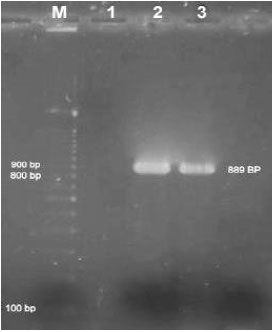 | |
| Fig. 4: | Agarose Gel electrophoretic analysis of PCR product of hlyEHEC gene of E.coli O157:H7. (Lane M: 100 bp DNA ladder, Lane 1: Negative control, Lane 2: hlyEHEC gene product (889 bp)) |
The results confirm amplification of ompW and hylEHEC genes in shrimp samples spiked with Vibrio and E.coli strains.
| Table 2: | Comparison of multiplex and single PCR for the detection of pathogens in spiked shrimp samples |
 | |
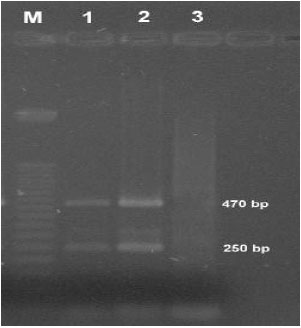 | |
| Fig. 5: | Agarose gel electrophoretic analysis of products of multiplex PCR (Lane M: 50 bp DNA ladder, Lane 1 and 2: Multiplex PCR product (470 bp confirms sefA and 250 bp confirms tdh) Lane 3: Negative control) |
Electrophoresis of multiplex PCR product of sefA and tdh genes: Multiplex PCR products were subjected to electrophoresis and two distinct PCR product bands were obtained and they were measured as 470 bp and 250 bp (Fig. 5). The results indicate that the designed multiplex set up was able to amplify sefA gene of Salmonella sp. and tdh gene of V. parahaemolyticus in a single reaction mixture.
Electrophoresis of multiplex PCR product of sefA, tdh, ompW and hlyEHEC genes: DNA samples extracted from spiked shrimp samples were investigated in multiplex PCR set up for simultaneous detection of all the target pathogens from shrimp sample. The PCR products were subjected to electrophoresis and results showed that all the four genes were amplified in multiplex set up (Fig. 6) as similar to that they were amplified in case of multiplex set up designed for tdh and sefA (Fig. 5).
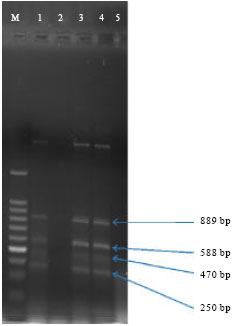 | |
| Fig. 6: | Agarose gel electrophoretic analysis of product of multiplex PCR set up for simultaneous detection of four target genes from spiked shrimp sample, Lane M: 100 bp ladder, Lane 1, 2, 3, 4 Multiplex PCR product: 470 bp confirms sefA, 250 bp confirms tdh, 588 bp confirms ompW and 889 bp confirms hlyEHEC Lane 5: Negative control |
Evaluation of multiplex PCR with spiked and pure DNA: For comparative analysis of uniplex and multiplex PCR set ups, results of different combinations uniplex and multiplex PCR has been summarized in Table 2. Uniplex amplifications yielded positive result in 70-80% spiked shrimp samples. Multiplex PCR setup with two genes yielded positive result in 75% of spiked shrimp samples whereas, multiplex PCR setup targeting four genes can detect the genes in 55% spiked shrimp samples.
DISCUSSION
Foods contaminated with pathogens play an important role in the dissemination of the pathogens to humans (Porteen et al., 2007) thus causes foodborne outbreak and concerns for public health. Development of PCR based modified detection methods are of huge interest because of more sensitivity, less time requirement and easy to handle large sample volume in the testing laboratories. In a previous study (Fakruddin et al., 2012), multiplex PCR method for simultaneous detection of Salmonella spp. and Vibrio parahaemolyticus from enrichment cultures and spiked shrimp amples were reported.. The objective of this study was to extend the method for simultaneous detection of four important food pathogens (Escherichia coli O157:H7, Salmonella sp., Vibrio parahaemolyticus and Vibrio cholerae) from shrimp samples and to compare the multiplex PCR performance with uniplex PCR results in terms of sensitivity and specificity. The targeted genes were ompW for V. cholerae, tdh for V. parahaemolyticus, hlyEHEC for E. coli O157:H7 and sefA for Salmonella sp.
All the four targeted genes were amplified successfully, mixed DNA and DNA extracted from co-culture were subjected to a number of multiplex settings designed to detect the pathogens simultaneously. In some of the multiplex settings there was no amplified product indicating the setting was not suitable for amplification. In 3 multiplex settings (multiplex setting 1-3), only two of the genes (tdh and sefA) were detected. The limitation of the PCR settings to detect hlyEHEC and ompW may be due to primer incompatibility, difference in annealing temperature and lack of suitable PCR mixture. Again, difference in annealing temperature and product length (899, 588, 470 and 250 bp) may also be factors for the failure to detect 4 genes at a time. A negative PCR results does not necessarily indicate that no template DNA was present in the sample. Inhibitory substances present in a sample may influence the outcome of the PCR by lowering or completely preventing the amplification (Lund and Madsen, 2006). A false-negative PCR result is a major concern to the food industry where PCR is being used for pathogen detection; therefore, internal standards or controls have to be included in the PCR to avoid false-negatives (Jones et al., 2000).
The presence of certain components in shrimp samples can prevent recovery of injured cells and may require a longer enrichment phase prior to PCR detection. Optimization of methods for detection of different pathogens is necessary, in terms of both the DNA extraction and PCR amplification steps. Furthermore, different pathogens may require different enrichment media and procedures. However, we have overcome some of these limitations through the use of a nonselective enrichment step followed by a multiplex PCR assay for simultaneous detection of these important pathogens, reducing the total analysis time without loss of sensitivity.
A total of 20 shrimp samples were collected and spiked for the study. Spiked DNA samples were subjected to both uniplex and multiplex PCR and results were recorded. Two multiplex setup was applied, one aiming to detect two genes (tdh and sefA) and another aiming detection of all four genes (tdh, sefA, ompW and hlyEHEC). PCR with spiked DNA showed considerable variation in result. In uniplex PCR, tdh was detected in 16 (80%) sample, whereas sefA was detected in 15 (75%), ompW was detected in 13 (65%), hlyEHEC was detected in 12 (60%) samples (Table 1). In multiplex PCR with spiked DNA targeting two gene (tdh and sefA) resulted 75% amplification of both genes on the other hand multiplex PCR targetting all four genes resulted amplification of 9 out of 20 samples (45%). Some non-specific products were also produced in this set up (Fig. 6), this may be due to non-specific binding of any primer to shrimp DNA. To estimate the specificity of the method, DNA from shrimps spiked with non-target pathogens such as Bacillus cereus, Shigella flexneri and Staphylococcus aureus were subjected to multiplex PCR and found no amplification, indicates the specificity of the multiplex PCR method.
Kim et al. (2007) has developed a multiplex PCR assay for simultaneous detection of five foodborne pathogen such as E. coli O157:H7, Salmonella, Staphylococcus aureus, Listeria monocytogenes and Vibrio parahaemolyticus. Kawasaki et al. (2009) developed another multiplex PCR method for detection of Salmonella spp., Listeria monocytogenes and E. coli O157:H7 from various types of artificially inoculated food samples including shrimp. Labreuche et al. (2012) developed a quantitative multiplex PCR method with an internal amplification control for simultaneous detection of Vibrio penaecida and Vibrio nigripulchritudo from shrimp samples with high specificity and sensitivity (200 CFU mL-1).
Development of multiplex PCR method with amplification of all targeted genes will be a valuable alternative screening and detection method for shrimp to be exported in foreign countries. Further investigation for amplification of ompW and hylEHEC genes as well as enhancement of sensitivity, specificity and generation of data for validation are required.
CONCLUSION
Based on the results, it can be concluded that the Multiplex PCR method targeted for two genes i.e. tdh and sefA is suitable for screening and detection of V. parahaemoliticus and Salmonella sp. from shrimp samples but before that proper validations of the method are required. On the other hand multiplex PCR method targeted for all four genes are yet to develop for screening and detection. Detail investigation and root cause analysis for failure are needed and further research to be adopted for development of the method with amplification of all genes with high sensitivity and specificity.
ACKNOWLEDGMENT
This research project was carried out under a special allocation project entitled "Development and Validation of Multiplex PCR Method for Rapid Detection of Pathogenic Microorganisms from Shrimp Value Chain of Bangladesh", funded by Ministry of Science and Technology, Government of the People’s Republic of Bangladesh.
REFERENCES
- Ahari, H., D. Shahbazzadeh and A. Misaghi, 2009. Selective amplification of SEA, SEB and SEC genes by multiplex PCR for rapid detection of Staphylococcus aureus. Pak. J. Nutr., 8: 1224-1228.
CrossRefDirect Link - Ahmed, S. and M.N. Anwar, 2007. Bacteriological assessment of value-added ready-to-cook/eat shrimps processed for export from Bangladesh following the guidelines of international standards. Bangladesh J. Microbiol., 24: 81-84.
Direct Link - AlHaj, N., N.S. Mariana, A.R. Raha and Z. Ishak, 2007. Simultaneous Detection of Enteropathogenic E. coli and shiga toxin-producing E. coli by polymerase chain reaction. J. Applied Sci., 7: 2531-2534.
CrossRefDirect Link - Fakruddin, M., S. Islam, M.M. Ahmed, A. Chowdhury and M.M. Hoque, 2012. Development of multiplex PCR (Polymerase Chain Reaction) method for detection of Salmonella spp. and Vibrio parahaemolyticus from shrimp samples of Bangladesh. Asian J. Biol. Sci., 5: 76-85.
CrossRefDirect Link - Goel, A.K., S. Ponmariappan, D.V. Kamboj and L. Singh, 2007. Single multiplex polymerase chain reaction for environmental surveillance of toxigenic-pathogenic O1 and non-O1 Vibrio cholera. Folia Microbiol., 52: 81-85.
PubMedDirect Link - Hara-Kudo, Y., K. Sugiyama, M. Nishibuchi, A. Chowdhury and J. Yatsuyanagi et al., 2003. Prevalence of pandemic thermostable direct hemolysin-producing Vibrio parahaemolyticus O3:K6 in seafood and the coastal environment in Japan. Applied Environ. Microbiol., 69: 3883-3891.
CrossRefDirect Link - Islam, M.A., A.E. Heuvelink, E. de Boer, P.D. Sturm and R.R. Beumer et al., 2007. Shiga toxin-producing Escherichia coli isolated from patients with diarrhoea in Bangladesh. J. Med. Microbiol., 56: 380-385.
CrossRef - Jofre, A., B. Martin, M. Garriga, M. Hugas, M. Pla, D. Rodrıguez-Lazaro and T. Aymerich, 2005. Simultaneous detection of Listeria monocytogenes and Salmonella by multiplex PCR in cooked ham. Food Microbiol., 22: 109-115.
CrossRef - Jones, R.N., M.L. Neale, B. Beattie, D. Westmoreland and J.D. Fox, 2000. Development and application of a PCR-based method including an internal control for diagnosis of congenital cytomegalovirus infection. J. Clin. Microbiol., 38: 1-6.
Direct Link - Kawasaki, S., N. Horikoshi, Y. Okada, K. Takeshita, T. Sameshima and S. Kawamoto, 2005. Multiplex PCR for simultaneous detection of Salmonella spp., Listeria monocytogenes and Escherichia coli O157:H7 in meat samples. J. Food Prot., 68: 551-556.
PubMedDirect Link - Kawasaki, S., P.M. Fratamico, N. Horikoshi, Y. Okada, K. Takeshita, T. Sameshima and S. Kawamoto, 2009. Evaluation of a multiplex PCR system for simultaneous detection of Salmonella spp., Listeria monocytogenes and Escherichia coli O157:H7 in foods and in food subjected to freezing. Foodborne Pathogens Dis., 6: 81-89.
CrossRefDirect Link - Khan, A.W., S.J. Hossain and S.N. Uddin, 2007. Isolation, identification and determination of antibiotic susceptibility of Vibrio parahaemolyticus from shrimp at Khulna region of Bangladesh. Res. J. Microbiol., 2: 216-227.
CrossRefDirect Link - Kim, J.S., G.G. Lee, J.S. Park, Y.H. Jung and H.S. Kwak et al., 2007. A novel multiplex PCR assay for rapid and simultaneous detection of five pathogenic bacteria: Escherichia coli O157:H7, Salmonella, Staphylococcus aureus, Listeria monocytogenes and Vibrio parahaemolyticus. J. Food Prot., 70: 1656-1662.
PubMedDirect Link - Labreuche, Y., L. Pallandre, D. Ansquer, J. Herlin, B. Wapotro and F. Le Roux, 2012. Pathotyping of Vibrio isolates by multiplex PCR reveals a risk of virulent strain spreading in new caledonian shrimp farms. Microb. Ecol., 63: 127-138.
CrossRefPubMedDirect Link - Lund, M. and M. Madsen, 2006. Strategies for the inclusion of an internal amplification control in conventional and real time PCR detection of Campylobacter spp. in chicken fecal samples. Mol. Cell. Probes, 20: 92-99.
PubMedDirect Link - Malkawi, H.I. and R. Gharaibeh, 2004. Rapid and simultaneous identification of two Salmonella enterica serotypes, enteritidis and typhimurium from chicken and meat products by multiplex PCR. Biotechnology, 3: 44-48.
CrossRefDirect Link - Malorny, B., P.T. Tassios, P. Radstrom, N. Cook, M. Wagner and J. Hoorfar, 2003. Standardization of diagnostic PCR for the detection of foodborne pathogens. Int. J. Food Microbiol., 83: 39-48.
CrossRefPubMedDirect Link - Mandal, P.K., A.K. Biswas, K. Choi and U.K. Pal, 2011. Methods for rapid detection of foodborne pathogens: An overview. Am. J. Food Technol., 6: 87-102.
CrossRefDirect Link - Oliveira, S.D., C.R. Rodenbusch, M.C. Ce, S.L.S. Rocha and C.W. Canal, 2003. Evaluation of selective and non-selective enrichment PCR procedures for Salmonella detection. Lett. Applied Microbiol., 36: 217-221.
CrossRefPubMedDirect Link - Porteen, K., R.K. Agarwal and K.N. Bhilegaonkar, 2007. Detection of Aeromonas sp. from chicken and fish samples by polymerase chain reaction. Am. J. Food Technol., 2: 30-37.
CrossRefDirect Link - Rijpens, N.P. and L.M. Herman, 2002. Molecular methods for identification and detection of bacterial food pathogens. J. AOAC. Int., 85: 984-995.
PubMed - Sahasraundhe, A. and M. Deodhar, 2010. Standardization of DNA extraction and optimization of RAPD-PCR conditions in Garcinia indica. Int. J. Bot., 6: 293-298.
Direct Link - Seidavi, A.R., S.Z. Mirhosseini, M. Shivazad, M. Chamani and A.A. Sadeghi, 2008. Optimizing multiplex polymerase chain reaction method for specific, sensitive and rapid detection of Salmonella sp., Escherichia coli and Bifidobacterium sp. in Chick gastrointestinal tract. AJAVA., 3: 230-235.
CrossRefDirect Link - Sherfi, S.A., I.E. Aradaib and H.A. Dirar, 2006. Evaluation of polymerase chain reaction for rapid detection of E. coli strains: A preliminary study. Asian J. Cell Biol., 1: 9-13.
CrossRefDirect Link - Yasmin, M., S. Kawasaki and S. Kawamoto, 2007. Evaluation of multiplex PCR system for simultaneous detection of Escherichia coli O157:H7, Listeria monocytogenes and Salmonella enteritidis in shrimp samples. Bangladesh J. Microbiol., 24: 42-46.
Direct Link - Yunus, M., 2009. EU Ban, HACCP compliance and shrimp exports from Bangladesh. Bangladesh Dev. Stud., Vol. 32. No. 3.
Direct Link