
Hamid Mir Mohammad Sadeghi
Department of Pharmaceutical Biotechnology and Isfahan Pharmaceutical Research Center, School of Pharmacy, Isfahan University of Medical Sciences and Health Services, Isfahan, Iran
Ali Jahanian Najafabadi
Department of Pharmaceutical Biotechnology and Isfahan Pharmaceutical Research Center, School of Pharmacy, Isfahan University of Medical Sciences and Health Services, Isfahan, Iran
Daryoush Abedi
Department of Pharmaceutical Biotechnology and Isfahan Pharmaceutical Research Center, School of Pharmacy, Isfahan University of Medical Sciences and Health Services, Isfahan, Iran
Abbas Jafarian Dehkordi
Department of Pharmaceutical Biotechnology and Isfahan Pharmaceutical Research Center, School of Pharmacy, Isfahan University of Medical Sciences and Health Services, Isfahan, Iran
Journal of Biological Sciences
Year: 2008 | Volume: 8 | Issue: 4 | Page No.: 826-830







ABSTRACT
In this study we used a nucleic acid based method to identify the capability of Polyhydroxy alkanoate (PHA) production of two isolates of Pseudomonas aeroginosa deposited in Persian Type Culture Collection (i.e., Pseudomonas aeroginosa PTCC 1310 and 1740) and present results showed this capability for the PTCC1310 but not for the other isolate. This identified isolate could be used for massive production of the mentioned polymers or its pha locus could be cloned and used for the functional expression of the PHA biopolymers. Here we also compared three bacterial genomic DNA extraction methods with respect to their difficulty, cost and the time of procedure. These methods included simple boiling method, phenol-chloroform method and commercial DNA extraction kit. All three methods gave reasonable amount of DNA that could be used as templates for the detection of pha gene. In conclusion we have identified a Pseudomonas aeroginosa strain present in Iran containing the necessary genes for PHA production and suggest that the simple boiling method can be utilized for further screening of bacterial samples for the presence of these genes.
PDF Abstract XML References Citation
How to cite this article
Hamid Mir Mohammad Sadeghi, Ali Jahanian Najafabadi, Daryoush Abedi and Abbas Jafarian Dehkordi, 2008. Identification of an Isolate of Pseudomonas aeroginosa Deposited in PTCC as a PHA Producer Strain: Comparison of Three Different Bacterial Genomic DNA Extraction Methods. Journal of Biological Sciences, 8: 826-830.
DOI: 10.3923/jbs.2008.826.830
URL: https://scialert.net/abstract/?doi=jbs.2008.826.830
DOI: 10.3923/jbs.2008.826.830
URL: https://scialert.net/abstract/?doi=jbs.2008.826.830
INTRODUCTION
The unlimited use of petroleum based plastics for different applications and purposes have created many problems related to the disposal of solid wastes (Kessler and Witholt, 1999; Salehizade and Van Loosdrecht, 2004). Therefore, worldwide programs have been started to solve the problems caused by the accumulation and non-decompository properties of these materials (Braunegg et al., 1998; Lee and Chang, 1995). One of the solutions to solve the problem is the use of biodegradable polymers, which will decompose after their disposal (Shimao, 2001). Among the candidates for biodegradable biopolymers, polyhydroxyalkanoates (PHAs) have received much attention because of their similar properties to synthetic plastics and their potential use as biodegradable and biocompatible thermoplastics (Aldor and Keasling, 2003; Salehizade and Van Loosdrecht, 2004) used for different industrial, marine, agricultural and also medical applications (Vidal-Mas et al., 2001; Agus et al., 2006a). Absorbable surgical suture, prosthesis or tissue engineering material and controlled release of drugs are some applications of these biopolymers in medicine (Zinn et al., 2001; Wang et al., 2003, Rossi et al., 2004).
PHAs are natural storage compounds which are mostly produced by a great number of microorganisms when in the presence of excessive carbon sources they encounter nutrient limitation. Some microorganisms, however, accumulate these polymers from the beginning of the exponential phase (Vidal-Mas et al., 2001; Agus et al., 2006b; Lemos et al., 2006). These biodegradable biopolymers are classified into three groups including short chain length (scl) PHAs containing 3 to 5 carbon atoms in monomer units, medium chain length (mcl) PHAs containing 6 to 14 carbon atoms in monomer units and scl-co-mcl containing 3 to 14 carbon atoms in monomer units (Solaiman and Ashby, 2005a).
PHA synthases are enzymes responsible for the polymerization and production of PHA. On the basis of structure, substrate specificity and organization of the gene locus these enzymes are classified into four classes. Various types of PHA producer microorganisms harbor different types of pha gene loci (Rehm, 2003). The class II pha operon consists of two PHA synthase genes (phaC1 and phaC2) flanking a PHA depolymerase gene (phaZ) and is found in mcl-PHA producing pseudomonas (Rehm, 2003; Solaiman and Ashby, 2005b). Because of the importance of the PHAs mentioned above, many groups developed various methods including lipophilic dye staining and nucleic acid based methods to identify the PHA producing microorganisms (Solaiman and Ashby, 2005b; Tyo et al., 2006). Among the nucleic acid based methods such as Southern blot hybridization based method developed by Timm et al. (1994) and PCR based methods developed by Lopez et al. (1997) or Solaiman et al. (2000), the later is highly specific for the identification of microorganisms harboring class II PHA polymerase. Since it has been shown that some Pseudomonas strains can produce these polymers, in the present study we focused on the identification of the PHA genes in Pseudomonas aeroginosa available in Persian Type Culture Collection (i.e., Pseudomonas aeroginosa PTCC 1310 and PTCC 1740).
To date various genomic DNA extraction methods developed and reported by different groups. The most important differences between these methods are the used reagents, the cost, time and the difficulty of procedure. In case of screening a large amount of samples a less expensive and easier method is preferred. In order to find such an optimal method for our later studies on the screening of other bacteria for their PHA production capability, here we also compared three different genomic DNA extraction methods including phenol-chloroform method, simple boiling method and extraction of genomic DNA using a commercial kit to clarify the advantages and disadvantages of each method against the others.
MATERIALS AND METHODS
Bacterial strains: Pseudomonas aeroginosa PTCC 1310 and PTCC 1740 were obtained from Persian Type Culture Collection (Tehran, Iran). As positive control Pseudomonas putida DSMZ 1693 and Pseudomonas oleovorance DSMZ 1049 were obtained from DSMZ, Germany. All the used bacteria were cultivated on nutrient agar or in nutrient broth before the extraction of genomic DNA.
Genomic DNA extraction: In this study three different methods of genomic DNA extraction were compared:
Simple boiling: In this method bacterial colonies were suspended in 1 mL TE buffer (1 mM EDTA, 10 mM Tris, pH = 8) and boiled for 10 min. Subsequently, the samples were transferred to -70 °C and kept for 5 min. Finally the samples were placed at 4 °C without any centrifugation for later use.
Phenol-chloroform method: The bacteria pelleted by centrifugation at 3500 rpm for 15 min. The supernatant was discarded and the pellet was resuspended in 200 μL of digestion buffer (100 mM Tric-Cl (pH 8.0), 5 mM EDTA (pH 8.0), 1% SDS) containing freshly thawed proteinase K (500 μg mL-1). The sample was then incubated at 55 °C in a water bath overnight. After the incubation, the tube content was mixed with an equal volume of phenol-chloroform-isoamyl alcohol solution (25:24:1), then centrifuged for 5 min at 13000 rpm. The supernatant was transferred into another clean tube carefully and without touching the pellet. Then, to remove the remained amount of phenol it was washed with the chloroform-isoamyl alcohol (24:1) solution. In the next step, 3 M sodium acetate was added to the mixture at a final concentration of 0.3 M. Two fold volume of ice-cold absolute ethanol was added and the content was centrifuged at 0 °C and 13000 rpm for 10 min resulting in a white precipitate. The upper liquid was discarded and 70 μL ethanol 70 °C was added to the tube and centrifuged for 5 min. The upper liquid was discarded and the precipitated DNA was dissolved in 200 μL double distilled water and kept at 4 °C for later use.
Commercial kit: Genomic DNA of the used strains was extracted using High Pure PCR template preparation kit (Roche, Germany) following the manufacturer`s instruction.
PCR: In order to screen the capability of PHA production, a PCR based method developed by Solaiman et al. (2000) was applied and the oligonucleotides 5´-ACA GAT CAA CAA GTT CTA CAT CTT CGAC-3´ and 5´-GGT GTT GTC GTT GTT CCA GTA GAG GAT GTC-3´ were used as suggested by that study. PCR condition included a primary denaturation time at 94 °C for 5 min, followed by 35 cycles of 1 min at 94 °C, 2 min at 55 °C and 3 min at 72 °C and a final extension time of 5 min at 72 °C. PCR reaction contained: 1 unit Taq DNA polymerase, 3 μL of 25 pM of each primer, 1 μL template DNA, 5 μL of 10 X PCR buffer and 1 μL dNTP (100 mM) at a final volume of 50 μL. The PCR products were examined on a 0.7% agarose gel containing a concentration of 0.5 μg mL-1 ethidium bromide to visualize the bands.
RESULTS
PCR amplification in order to identify the capability of PHA production: Genomic DNA of P. putida (DSMZ 1693) and P. oleovorans as positive controls and also genomic DNA of P. aeroginosa PTCC 1310 and PTCC 1740 were extracted using commercial kit and examined by agarose gel electrophoresis (figure not shown). All the extracted genomes were used as templates for PCR reaction.
In order to screen the existence of type II pha gene locus a primer pair (see Materials and Methods) was used for PCR analysis. A band at about 540 bp was expected if this locus was present. As shown in Fig. 1, gel electrophoresis of the PCR products for P. putida (DSMZ 1693), P. oleovorans (DSMZ 1049) and P. aeroginosa PTCC1310 but not P. aeroginosa 1740 gave the expected band and these results were reproducible. For more examination, the PCR reaction for P. aeroginosa PTCC 1740 was performed using different concentration of MgCl2 ranging from 2 to 4 mM or under different annealing temperature ranging from 52 to 60 °C but the desired band was not observed (data not shown).
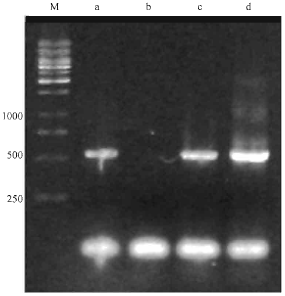 |
| Fig. 1: | Gel electrophoresis of PCR products using genomic DNA extracted by commercial kit as PCR template. As it shows the isolate P. aeroginosa 1310 and the two positive controls including P. putida DSMZ 1693 and P. Oleovorans DSMZ 1049 revealed the expected band at about 540 bp region (lanes a, c and d respectively). But the PCR amplification reaction for the isolate P. aeroginosa PTCC1740 resulted in no product. M represents molecular size marker. Two microliter of each sample was run |
Comparison of different genomic DNA extraction methods: Having proved the existence of pha locus in genomic DNA of P. aeroginosa PTCC 1310, different genomic DNA extraction methods were performed to extract its genomic DNA. In fact, commercial kit, simple boiling method and phenol-chloroform method were performed to extract genomic DNA (see materials and methods) and the extracted products were gel electrophoresed (Fig. 2).
 |
| Fig. 2: | Genomic DNA extraction of P. aeroginosa PTCC1310 by using different methods including (a) commercial kit, (b) simple boiling and (c) phenol-chloroform method. Two microliter of each sample was electrophoresed |
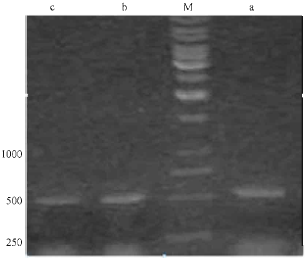 |
| Fig. 3: | Gel electrophoresis of PCR products using genomic DNA extracted by (a) commercial kit, (b) phenol-chloroform and (c) simple boiling methods. As it shows when DNA template obtained from these methods were used, the obtained PCR products were similar in size with almost equal intensity and sharpness. M represents DNA size marker. Two microliter of each sample was electrophoresed |
In order to examine the efficiency of extraction methods, extracted DNA was used as template for PCR amplification using the mentioned primers. Gel electrophoresis of the PCR products revealed same bands at about 540 bp for all three DNA samples (Fig. 3).
DISCUSSION
To date different methods have been used to screen the existence of pha locus in various microorganisms and also the capability of PHA production by them. Lipophilic dyes such as sudan black B, Nile blue A and Nile red have been traditionally used for the screening of PHA containing bacteria (Spiekermann et al., 1999; Gorenflo et al., 1999; Vidal-Mas et al., 2001). However, because of their non specific lipid binding property, these dyes could also stain other lipid materials. Because of this non specific staining property and some other problems such as being time consuming and the possibility of false negative results which would occur when a bacterium harbor the genetic organization for PHA production but the condition is not suitable to produce it, several nucleic acid based methods were developed by different groups like Timm et al. (1994), Lopez et al. (1997) and Solaiman et al. (2000); the latest group developed the first PCR procedure that specifically detects class II pha genes. They used a pair of primers designed on the basis of a highly conserved region of class II pha loci of various Pseudomonas. In this study, we used the primer pair suggested by Solaiman et al. (2000) as we examined the PHA production of two Pseudomonas aeroginosa isolates. Gel electrophoresis of the PCR products revealed the expected bands for the positive controls (P. putida DSMZ 1693 And P. oleovorans DSMZ 1049) and also for P. aeroginosa PTCC 1310 but not for P. aeroginosa 1740 confirming the capability of P. aeroginosa PTCC 1310 to produce medium chain length polyhydroxyalaknoates.
Here we also compared three different genomic DNA extraction methods. The comparison was based on different criteria like accuracy, difficulties, time and cost of the experiment and reproducibility of results. Commercial kits, despite their fast and accurate procedure are expensive. In comparison phenol-chloroform method is less expensive and also accurate, but it is time consuming and the procedure should be performed under specific condition to prevent phenol oxidation and also chloroform evaporation. Simple boiling method revealed reasonable results. This method is fast, inexpensive, repeatable and also does not need any specific reagents. But the most important problem of this method is limited time of keeping samples for later use. In this method DNA is not separated from cell lysates and other cytoplasmic and membrane components which could destruct the genomic DNA. Therefore, beside the easy, inexpensive and fast properties of this method, further modifications are required.
In conclusion, in this study we identified the Pseudomonas aeroginosa PTCC1310 as a PHA producer microorganism which could be used in later studies for the production of the precious PHA materials, or for the cloning and molecular study of its pha gene locus in the expression host for the over production of PHA. Here we also found the simple boiling method as a reliable, reproducible, inexpensive and fast method for the extraction of bacterial genomic DNA in order to screen a large amount of bacteria to identify their PHA production capability on the basis of the mentioned PCR based method.
ACKNOWLEDGMENT
This study was supported by a research grant awarded by the Isfahn University of Medical Sciences. The authors would like to thank Ms. Fatemeh Moazen for her assistance in this research.
REFERENCES
- Agus, J., P. Kahar, H. Abe, Y. Doi and T. Tsuge, 2006. Molecular weight characterization of poly [(R)-3-hydroxybutyrate] sunthesized by genetically engineered strains of Escherichia coli. Polym. Degrad. Stabil., 91: 1138-1146.
Direct Link - Agus, J., P. Kahar, H. Abe, Y. Doi and T. Tsuge, 2006. Altered Expression of polyhydroxyalkanoate synthase gene and its effect on poly [(R)-3-hydroxybutyrate] synthesis in recombinant Escherichia coli. Polym. Degrad. Stabil., 91: 1645-1650.
Direct Link - Aldor, I.S. and J.D. Keasling, 2003. Process design for microbial plastic factories: Methabolic engineering of polyhydroxyalkanoates. Curr. Opin. Biotechnol., 14: 475-483.
CrossRef - Braunegg, G., G. Lefebvre and K.F. Genser, 1998. Polyhydroxyalkanoates, biopolyesters from renewable resources: Physiological and engineering aspects. J. Biotechnol., 65: 127-161.
PubMedDirect Link - Gorenflo, V., A. Steinbüchel, S. Marose, M. Rieseberg and T. Scheper, 1999. Quantification of bacterial polyhydroxyalkanoic acids by Nile red staining. Applied Microbiol. Biotechnol., 51: 765-772.
CrossRefDirect Link - Lee, S.Y. and H.N. Chang, 1995. Production of poly (hydroxyalkanoic acid). Adv. Biochem. Eng. Biotechnol., 52: 27-57.
CrossRefDirect Link - Lemos, P.C., L.S. Serafim and M.A.M. Reis, 2006. Synthesis of polyhydroxyalkanoates from different short-chain fatty acids by mixed cultures submitted to aerobic dynamic feeding. J. Biotechnol., 122: 226-238.
PubMedDirect Link - Lopez, N.I., M.J. Pettinari and B.S. Mendez, 1997. Detection of reserve polymer synthesis genes in natural bacterial populations. FEMS Microbiol. Ecol., 22: 129-136.
Direct Link - Rehm, B.H.A., 2003. Polyester synthases: Natural catalysts for plastics. Biochem. J., 376: 15-33.
Direct Link - Rossi, S., A.O. Azghani and A. Omri, 2004. Antimicrobial efficacy of a new antibiotic-loaded poly(hydroxybutyric-co-hydroxyvaleric acid) controlled release system. J. Antimicrob. Chemother., 54: 1013-1018.
Direct Link - Salehizade, H. and M.C.M. Van Loosdrecht, 2004. Production of polyhydroxyalkanoates by mixed culture: recent trends and biotechnological importance. Biotechnol. Adv., 22: 261-279.
CrossRef - Solaiman, D.K.Y., R.D. Ashby and T.A. Foglia, 2000. Rapid and specific identification of medium-chain-length polyhydroxyalkanoate synthase gene by polymerase chain reaction. Applied Microbiol. Biotechnol., 53: 690-694.
PubMedDirect Link - Solaiman, D.K.Y. and R.D. Ashby, 2005. Genetic characterization of the poly(hydroxyalkanoate) synthases of various Pseudomonas oleovorans strains. Curr. Microbiol., 50: 329-333.
CrossRefDirect Link - Solaiman, D.K.Y. and R.D. Ashby, 2005. Rapid genetic characterization of poly(hydroxyalkanoate) synthase and its applications. Biomacromology, 6: 532-537.
CrossRefDirect Link - Spiekermann, P., B.H.A. Rehm, R. Kalscheuer, D. Baumeister and A. Steinbuchel, 1999. A sensitive, viabale-colony staining method using nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch. Microbiol., 171: 73-80.
PubMedDirect Link - Timm, A., S. Wiese and A. Steinbuchel, 1994. A general method for identification of polyhydroxyalkanoic acid synthase genes from pseudomonads belonging to the rRNA homology group I. Applied Microbiol. Biotechnol., 40: 669-675.
CrossRefDirect Link - Tyo, K.E., H. Zhou and G.N. Stephanopoulos, 2006. High-throughput screen for poly-3-hydroxybutyrate in Escherichia coli and Synechocystis sp. strain PCC6803. Applied Environ. Microbiol., 72: 3412-3417.
CrossRefDirect Link - Vidal-Mas, J., O. Resina-Pelfort, E. Haba, J. Comas, A. Manresa and J. Vives-Rego, 2001. Rapid flow cytometry- Nile red assessment of PHA cellular content and heterogenicity in cultures of Pseudomonas aeroginosa 47T2 (NCIB 40044) grown in waste frying oil. Antonie van Leeuwenhoek, 80: 57-63.
CrossRefDirect Link - Wang, Z., Y. Itoh, Y. Hosaka, I.K. Ashi, Y. Nakano, I. Maeda, F. Umeda, J. Yamakawa, M. Kawase and K. Yag, 2003. Novel transdermal drug delivery system with polyhydroxyalkanoate and starburst polyamidoamine dendrimer. J. Biosci. Bioeng., 95: 541-543.
CrossRef - Zinn, M., B. Witholt and T. Egli, 2001. Occurrence, synthesis and medical application of bacterial polyhydroxyalkanoate. Adv. Drug Delivery Rev., 53: 5-21.
CrossRef