
Amgad Kamal
Valeo InterBranch Automative Software, Smart Village, Cairo, Egypt
Mohsen Mahroos
Department of Electronics and Electrical Communications, Cairo University, Giza, Egypt
Ahmed Sayed
Varkon Semiconductors, Cairo, Egypt
Amin Nassar
Department of Electronics and Electrical Communications, Cairo University, Giza, Egypt
Information Technology Journal
Year: 2012 | Volume: 11 | Issue: 8 | Page No.: 998-1006







ABSTRACT
Sequence alignment has become a fundamental process in computational biology as it helps in finding similarity regions between biological sequences that may indicate the common properties across the sequences. Global Multiple Sequence Alignment (MSA) is a way to align the entire group of biological sequences. The exact solution of the global MSA is an NP-complete problem and iterative sequence alignment is among the main heuristic methods used for solving this computationally expensive problem. This approach realigns and evaluates initial biological sequences repeatedly through a number of iterations. The iterative approach is mostly used in conjunction with other computational optimization approaches and the total number of iterations as well as the iteration time mainly affects the overall alignment time. In this study, a parallel Particle Swarm Optimization (PSO) algorithm is presented for solving the global MSA problem based on iterative sequence alignment. The algorithm has been implemented using the massage-passing interface (MPI) library and tested over a Linux cluster and over the EUMed Grid. Experimental results are presented that demonstrate the performance of the proposed algorithm using different proteins from the SABmark and BAliBASE benchmark databases, different substitution matrices and different gap penalty models.
PDF Abstract XML References Citation
Received: November 05, 2011;
Accepted: March 16, 2012;
Published: June 05, 2012
How to cite this article
Amgad Kamal, Mohsen Mahroos, Ahmed Sayed and Amin Nassar, 2012. Parallel Particle Swarm Optimization for Global Multiple Sequence Alignment. Information Technology Journal, 11: 998-1006.
DOI: 10.3923/itj.2012.998.1006
URL: https://scialert.net/abstract/?doi=itj.2012.998.1006
DOI: 10.3923/itj.2012.998.1006
URL: https://scialert.net/abstract/?doi=itj.2012.998.1006
INTRODUCTION
Sequence alignment is the procedure by which an attempt is made to infer which positions (sites or regions) within biological sequences are homologous, that is, which sites share a common evolutionary history (Rosenberg, 2009). When DNA (deoxyribonucleic acid), RNA (ribonucleic acid), or protein sequences are aligned, new gaps are inserted in the middle of the sequences so that homologous sites are identified but there is no change in the amino acids order.
Sequence alignment is generally classified into two types; global alignment and local alignment. Entire sequences are considered in the former type, while only proper subsets are considered in the latter type. Global alignment is used when a whole species is to be characterized and when sequences are similar in length. Local alignment is used when a specific trait or property is sought and when sequences are dissimilar in length. Pairwise sequence alignment and more generally multiple sequence alignment (MSA) in which a group of two and more than two biological sequences, respectively, are aligned are fundamental for biological research and applications. For example, using sequence alignment has been reported for sequence analysis (Tambunan et al., 2007; Nur Fariza et al., 2008; Qasem et al., 2010; Hassanain et al., 2011), in silico (Rahim, 2010; Opabode et al., 2011), gene expression (Joshua et al., 2010), phylogenic tree construction and analysis (EI-Kholy et al., 2005; Kuracha et al., 2006; Suneetha et al., 2008; Elkalamawy et al., 2011) and molecular characterization (Elbeshehy and Sallam, 2012).
In this study, an iterative parallel Particle Swarm Optimization (PSO) algorithm is presented for solving the global MSA problem. The fundamentals of the sequence alignment are described and recent developments in using parallel computing for sequence alignment are discussed. The experimental results for the performance of the proposed algorithm are reported and analyzed.
SEQUENCE ALIGNMENT
Here, the fundamentals of the sequence alignment are described. The section starts with general classifications for sequence alignment algorithms. This is followed by an introduction of the concept of alignment score and the gap penalty model. The section ends with a review on recent progress in using parallel computing for sequence alignment.
Algorithm classification: There are several classifications for any sequence alignment algorithm. Each algorithm may fall under one or more of the following categories: Exact, progressive, or iterative algorithms and block-based, consistency-based and/or heuristic based (Mohsen and Abdullah, 2011).
Exact algorithms can provide optimum solutions but take long processing times and need high memory resources. The memory required to align two sequences of length m and n is O(mn). The maximum number of sequences that can be aligned simultaneously using this method is less than 20 (Notredame, 2002). The exact MSA problem is known to be NP-complete and is not feasible to be solved in reasonable processing time with the continuous growth of the biological sequence databases.
One of approximate methods for sequence alignment is the progressive method; which performs alignment on two sequences. The output is then aligned with a new sequence and the cycle continues till all sequences are aligned. Although this method is efficient but it has a drawback that there is no way to refine the errors in the initial alignments and hence these errors may propagate and increase in the subsequent cycles (Salam et al., 2005). This results in the method reaching a local optima and getting stuck there.
Iterative methods work similarly to progressive methods, but repeatedly realign the initial sequences and are mostly used in conjunction with other methods. New randomly generated or mutated sequences are added to the set of sequences, hence avoiding getting stuck in local optima. Iterative methods usually rely on stochastic algorithms, such as Genetic Algorithms GAs, that optimize a scoring function to search for the best alignment. This scoring function produces a metric that reflects biological events and optimizing this score leads to a correct alignment (Da Silva et al., 2011).
Consistency based algorithms focuses on reducing early alignment errors to avoid refinement stage for the final alignment. Block-based algorithms produce the alignment on two stages. In the first stage, they try to identity the conserved regions (blocks) that have no change in the amino acids. Then in the second stage, the regions between the successive blocks are aligned to produce the final alignment (Mohsen and Abdullah, 2011).
Progressive and iterative methods usually employ heuristic optimization approaches that work on optimizing an objective function and try to improve a candidate solution. This approach includes Genetic Algorithm, Ant Colony, Swarm Intelligence and Simulating Annealing. Examples of the heuristic algorithms that are presented in the literature recently include Liu et al. (2007), Da Silva et al. (2008) and Joo et al. (2008).
Alignment score: A group of sequences may be aligned in many different ways. Consequently, it is mandatory to evaluate these alignments so as to determine the best possible alignment. One of the evaluation methods is to calculate the total alignment score using a substitution matrix (scoring matrix). The substitution matrix describes the probability that amino acid changes to another one. It represents implicitly a particular theory of evolution. Scoring matrices appear in all analysis involving sequence comparison. The available scoring matrices include PAM (Point Accepted Mutation) (Dayhoff et al., 1978), BLOSUM (Block Substitution Matrix) (Henikoff and Henikoff, 1992) and many other matrices.
There are also different schemes to calculate the total alignment score. Two schemes are used in this research work: The Sum-of-Pairs Score (SPS) and the Identity Score (IS). SPS represents the sum of scoring that each sequence has with all other sequences using a substitution matrix and is more of a relative metric among groups of sequences. IS represents the sum of the number of matched characters between each two sequences and is perceived as an absolute metric.
Gap penalty model: As mentioned earlier, the amino acid residues are kept as they are when two sequences are aligned, but shifted as gaps are inserted in the middle between the residues. Gaps account for some evolutionary events that caused one character or more to be misaligned and/or mutated among sequences.
Having the least number of Gaps is the ultimate goal during the alignment of sequences. Due to this misalignment, a gap is weighted as a penalty during the alignment. Three models are currently used to evaluate the effect of gaps inserted within a sequence:
| • | Constant gap model: In this model a negative score is added to the alignment score whenever a gap is opened |
| • | Linear gap model: This model has only one parameter (d) which is the penalty per unit length of gap. This is always negative, so that the alignment with fewer gaps is favored over the alignment with more gaps. Under a linear gap penalty model, the overall penalty for one large gap is the same as for many small gaps |
| • | Affine gap model: The affine gap model penalizes insertions and deletions using a linear function in which one term is length-independent (Gapopen) while the other is length-dependent (Gaplength x Gapextend) |
| (1) |
The affine gap penalty model is considered more appropriate for aligning DNA and protein sequences (Chao and Zhang, 2009). This model is based on the notion that having gaps grouped together is more likely what happens during biological evolutionary events, resulting in better alignment among closely related sequences than having gaps widely distributed. When Gapopen is higher than Gapextend, this model favors extending the gap length over opening a new gap.
Recent Progress: Biological databases have recently grown to humongous sizes and are still in continual growth. This led the global multiple sequence alignment problem to become increasingly expensive in terms of the required memory resources and alignment time. Researchers have made a lot of effort to find new and efficient solutions for this problem. Consequently, many algorithms and platforms have been developed and reported recently in the literature. The central idea in these solutions is to employ parallel computing to be able to obtain the results within an acceptable time with sufficient accuracy. The reported solutions can be classified into hardware specific solutions and software specific solutions.
Hardware solutions depend on new developed hardware platforms and architectures and try to reduce the alignment time by executing different parts of the algorithm in parallel using the available hardware resources. The recent solutions include FPGA-based reconfigurable hardware platforms, Graphics Processing Units (GPU) (Ligowski and Rudnicki, 2009), Cell BE from IBM (Sarje and Aluru, 2008), general purpose Multi-Core processors (De Almeida and Roma, 2010) and Network-on-Chip (NoC) platforms (Sarkar et al., 2010).
Software solutions depend on the libraries and applications programming interfaces (API) as they usually target the general-purpose platforms. These APIs include new parallel programming techniques like Pthreads (from IEEE Portable Operating System Interface POSIX), OpenMP (Open Multi-Processing) for shared memory platforms and MPI library for distributed memory processing. New paradigms are trying to cross this gap between software and hardware; for example OpenCL framework (Lee et al., 2010), CUDA (Luebke, 2008), MultiProcessor Java (Nordin and Rahman, 2009) and even parallel programming languages like Cilk (Joerg, 1996), Brook (Buck et al., 2004), NESL (Blelloch, 1995), ZPL (Chamberlain et al., 1998) and UPC (UPC Consortium, 2005).
In the past few years, software solutions used the parallel programming intensively to implement different multiple sequence alignment tools and algorithms using the MPI library and target cluster computing. This is due to the ability of parallel programming to achieve good scalability and speedup without affecting the accuracy as reported in different studies (Li, 2003; Boukerche et al., 2007).
PARALLEL PSO ALGORITHM
The success of the iterative PSO algorithm reported (Rodriguez et al., 2007) in improving the accuracy of the initial alignment generated by the Culstal X MSA tool motivated the development of the parallel PSO algorithm presented in this section which is a novel parallelization of the algorithm presented by Rodriguez et al. (2007) with modifications in the crossover point determination.
The iterative PSO algorithm is a population-based evolutionary algorithm that was inspired by the social behavior of birds flocking or fish schooling. The PSO algorithm is suitable for solving the alignment problem as it can be used to realign and evaluate the initial alignment to achieve more accurate results. In addition to that, it can support different objectives functions. So, the final alignment can be changed according to the need. For example, it may be desired in some cases to find the regions of high number of matched residues. In such a case, the objective function may be chosen to maximize the identity score.
In the iterative PSO algorithm, a set of initial solutions called particles are continuously improved in an iterative approach until an acceptable solution is met. In each iteration, particles are evaluated using a fitness function and the most fit particle is selected as the new leader. This fitness function represents the solution to the problem that should be solved. After selecting the leader particle, each particle moves towards the leader in a speed proportional to the distance between the leader and the particle.
In every iteration of the PSO algorithm, one (or more) random crossover point(s) is selected based on that distance and a mutation occurs with the current leader, resulting in the swarm moving towards the leader at different speeds without getting stuck in a local optimal solution point. The goal of iterative methods is to explore the search space and improve the alignment results.
Though the direct approach on parallelizing the iterative PSO algorithm on cluster computing platforms partitions the data over the cluster to improve the speedup, this approach has several drawbacks. First, it limits the search scope of each node which would most likely affect the quality of overall results. Second, it has a high communications cost among nodes due to the nature of the iterative PSO algorithm which results in scalability issues, load balancing (or task mapping) problem among nodes with different sequences of different lengths and the overhead of distributing the work over the cluster.
The proposed parallel PSO algorithm is an optimized parallel implementation of the iterative PSO algorithm using the MPI library that enables running simultaneously in parallel pieces of the large sequential iterative PSO algorithm. This proposed parallel approach led to some modifications in the design of the PSO algorithm. A Linux cluster with configurable computational nodes is used as the running environment for parallel computing. The objective function of the PSO problem is to maximize the SPS score or the identity score. Each particle represents a possible alignment and it contains all the sequences. First, an initial alignment is obtained using any alignment tool like Clustal X (Jeanmougin et al., 1998). This alignment is then used as the input to the PSO algorithm and used to generate the other particles by making random space insertions in this initial alignment. The number of spaces to be inserted is the user’s choice and when it is set to zero, the original spaces are changed randomly so that they have new positions. The proposed parallel PSO algorithm is shown in Fig. 1.
When sequences are aligned, the amino acid residues in each sequence are kept in the same order, but are shifted so that homologous sites are in the same positions as in the aligned sequences. This means that sequences in all particles are similar in the residues, but are different in the positions of gaps and there is no need to save the whole sequence. Only gap positions are saved for each sequence. Theses gaps form a matrix as shown in Fig. 2, where each row represents the gaps saved in one sequence.
To have a load-balanced parallel processing, the total number of particles is divided over the available processes in the parallel computing system. Each process in such system has its own particles and works on them independently. Also, each process stores a copy of the current leader particle. Thus at the end of each iteration, all processes communicate to determine the new leader and exchange its sequences. The data representation is shown in Fig. 3 for the proposed parallelization of the iterative PSO algorithm with K sequences, m processes, n particles.
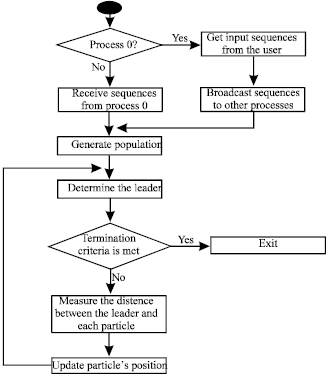 | |
| Fig. 1: | Proposed parallel PSO algorithm |
 | |
| Fig. 2: | Data saved per particle |
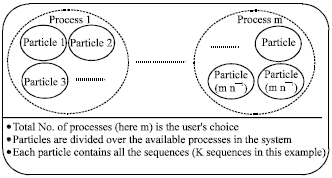 | |
| Fig. 3: | Data representation for the PSO algorithm |
The particles are evaluated each iteration using either the SPS objective function or the IS objective functions and a new leader particle based on this metric is chosen. The mathematical formulas of the two objectives functions are illustrated below:
| (2) |
| (3) |
Leader determination is performed in two steps. In the first step, each process scans its own particles to determine the local leader. Then in the second step, processes communicate together to determine the global leader. This mechanism happens as follows. Each process broadcasts its ID and local leader best score to all other processes and by comparing the other processes’ scores, the global leader process is determined and it begins to broadcast the leader sequences to the other processes. When the number of processes is small, the described broadcast algorithm is efficient and there is no need to use a divide-and-conquer approach for leader determination. The leader determination process is shown in Fig. 4. After determining the leader, the other particles have to move towards the leader in a speed proportional to the current distance between the particle and the leader. This distance is measured as follows:
| (4) |
For example, the distance between the particle and the leader for the two particles in Fig. 2 is calculated as follows:
| • | The total number of gaps is 12 |
| • | Number of unmatched gaps are 4 and they are (6,7) and (8,7) |
Therefore, the distance is 4/12 = 0.333. The movement towards the leader takes effect by replacing a part from the particle’s sequence by another part from the leader’s sequence. Double crossover points are used to determine the parts of the sequence that should be replaced as follows:
| • | Calculate the distance between the leader and the particle |
| • | Determine the length of the fixed part (L) that will not be replaced by the leader sequence as follows: |
| • | Replace the whole particle’s sequence by the leader’s sequence except for the part of length L that begins at a random point within the sequence length. The flowchart used to determine the crossover point is shown in Fig. 5 |
| (5) |
| (6) |
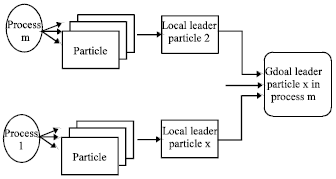 | |
| Fig. 4: | Determining the leader |
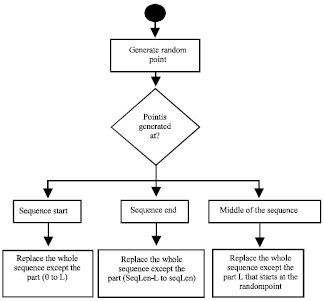 | |
| Fig. 5: | Determining the crossover points |
Due to the replacement, the sequences may become longer or shorter. Hence, some adaptation is needed each iteration to equalize the length of all particles. It may happen also that a column of spaces is generated in all sequence. This column is useless and not considered in the calculation of SPS score.
The proposed PSO algorithm has some configuration parameters that affect the final alignment accuracy and alignment time. These parameters include the population size (number of particles), number of iterations, gap penalties, number of inserted spaces, the scoring matrix and the number of parallel processes. There is tradeoff between accuracy and alignment time for some of these parameters. For example, increasing the population size increases both the algorithm accuracy and alignment time.
EXPERIMENTAL RESULTS
In this study, the proposed parallel PSO algorithm was developed using the MPICH2 (Gropp, 2002), an implementation of the MPI library. The Amazon Elastic Compute Cloud (Akioka and Muraoka, 2010) was used to create a High-Performance Computing (HPC) Linux cluster with configurable number of computational instances for executing the proposed parallel PSO algorithm and investigating its accuracy and scalability.
The algorithm was also tested on the EUMed Grid (Andronico et al., 2007). It was possible to increase the number of used particles in the tests via utilizing the available resources on the Grid. In this section, a comparative analysis between the EUMed Grid results and the Linux cluster results is presented.
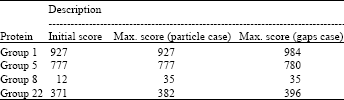
Accuracy results: Accuracy tests were done using different protein families from SABmark (Van Walle et al., 2005) and BAliBASE (Thompson et al., 1999) databases. SABmark currently contains two sets, each consisting of a number of subsets with related sequences. The first set, the Twilight Zone set, contains sequence with very low similarity, between 0-25% identity and a common evolutionary origin cannot be established between most pairs even though their structures are (distantly) similar. This set therefore represents the worst case scenario for sequence alignment. The second set, the Superfamilies set, contains sequences with a (putative) common evolutionary origin. However, they share at most 50% identity which is still challenging for any sequence alignment algorithm. Two protein families were used from each set to test the proposed algorithm, as described in Table 1.
The different protein families are firstly aligned using Clustal X. Then, the Clustal X’s output is used as an input to the proposed parallel PSO algorithm. In order to cover much of the alignment search space for each protein family, two types of tests were performed. The first test is to start with 125 particles and successively double the number of particles until it reaches 16000. The second test is to keep the number of particles constant at 8000, but to start with 1 inserted gap and successively double the number of gaps until it reaches 128. In each of these two tests, the test is repeated 15 times and the average score and maximum score values are calculated, so as to obtain statistically sound results.
To evaluate the algorithm under different gap penalty models and substitution matrices, the above tests are repeated to cover the following four cases: PAM250 with affine gap model, PAM250 with constant gap model, BLOSUM45 with affine gap model and BLOSUM45 with constant gap penalty model.
| Table 1: | Used protein families description |
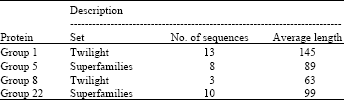 | |
| Table 2: | Maximum improvement achieved using the affine model by changing the number of particles from 125 to 16000 |
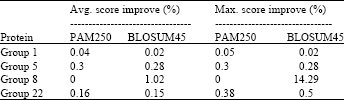 | |
| Table 3: | Maximum improvement achieved using the affine model by changing the number of inserted gaps from 1 to 128 |
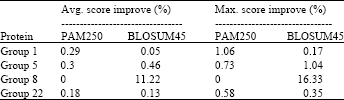 | |
The PSO configuration parameters were chosen as follows:
| • | Number of iteration = 100 |
| • | Gap open = 10 |
| • | Gap extension = 2 for affine gap model |
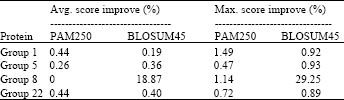
Table 2 and 3 show the maximum improvement obtained over all the experiments in case of SABmark database, affine gap model and SPS objective function, regardless of the number of particles and the number of new inserted gaps. From these results, it was observed that proposed algorithm improved the initial alignment in most cases. It was also observed that adding new spaces had better effect on the alignment score as expected. When no spaces were inserted (only changing the number of particles), the algorithm generates the particles by changing the sequences’ original spaces positions. If the initial sequences in this case are well aligned, then the amount of improvement that could be obtained will be small. Adding new spaces in the middle allows the characters to change their positions more and hence there is a better chance to be more aligned. The algorithm also gave almost similar results for the two substitution matrices used. This is attributed to the fact that PAM250 and BLOSUM45 matrices are almost equivalent.
Table 4 and 5 show the maximum improvement obtained over all the experiments regardless of the population size and inserted spaces with constant gap model and SPS objective function.
| Table 4: | Maximum improvement achieved using the constant gap model by changing the number of particles from 125 to 16000 |
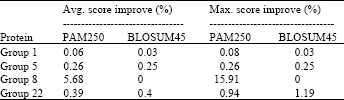 | |
| Table 5: | Maximum improvement achieved using the constant gap model by changing the number of inserted gaps from 1 to 128 |
 | |
| Table 6: | Maximum improvement achieved using the identity score by changing the number of particles from 125 to 16000 and the number of spaces from 1 to 128 |
 | |
From these results, it was observed that the constant gap model achieved higher alignment score. This is because affine gap model encourages grouping the gaps rather than adding many small gaps like the constant model. So, when the gaps are separated, there is more chance to shift each character separately to achieve higher score. In some cases, this may not be preferred, as it makes it hard to get the sequence properties when compared to another sequence. Also, although the constant gap model achieved higher score, the alignment length was increased by about 9%.
Table 6 shows the maximum improvement achieved using the identity score by changing the number of particles from 125 to 16000 and the number of spaces from 1 to 128. From these results, it was observed that the proposed algorithm improved the initial score and that spaces insertion had more effect than the number of the particles. This is due to the fact that if the initial alignment is good, it is hard to improve the alignment by just changing the original gaps’ positions (particle case) and spaces insertion is needed to achieve a better score.
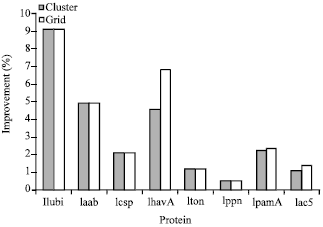
As mentioned above, the algorithm was also tested on the EUMed grid using different protein families from the BAliBASE benchmark. By using the available resources on the Grid, it was possible to increase the number of particles to 32000. A comparative analysis between the EUMed Grid results and the HPC Linux cluster results is shown in Fig. 6 when the Identity score objective function is used.
 | |
| Fig. 6: | EUMed grid results vs. Linux cluster results |
As shown in Fig. 6, the accuracy of the alignment is increased when the number of particles is increased, as the search space is increased and a better chance exists that a more aligned particle is added to the swarm. It is also observed that for some proteins, the algorithm reaches its maximum possible final alignment score by using the 16000 particles available on the cluster. Increasing the number of particles beyond this level by using the EUMed Grid has no effect on increasing the final alignment score which saturates to this maximum value.
Scalability analysis: To evaluate the algorithm performance and scalability, different types of tests were performed. In those tests, large sequences were used (up to 10000 residues). Also the number of used processes was changed to cover the range (1, 2, 4, 8, 16, 32) and the number of cluster instances was changed from 1 instance up to 6 instances. When one cluster instance is used with one process, this means that the algorithm is totally sequential and no parallelization at all.
It was observed that the algorithm speedup increases till the total number of the used processes on each cluster instance is 8, then the speedup begins to decrease again due to the overloading and context switching between the processes.
The algorithm was able to achieve a max speedup of 30 when 32 processes were used over 6 cluster instances comparing to the sequential case. In this case the algorithm time was reduced from 24 min to 0.8 min.
CONCLUSION
In this study, a parallel PSO algorithm is presented to solve the sequence alignment problem. The proposed algorithm was implemented using the MPICH2 and run over a Linux cluster. Algorithm accuracy was tested over 4 protein families using Clustal X’s output as an initial alignment. The proposed algorithm is not limited to the Clustal X tool and can work with any other tool for initial alignment.
It was observed that the algorithm improved the initial alignment score for all protein families under test. The proposed algorithm gave very similar results when tested using the PAM250 and BLOSUM45 substitution matrices. For gap penalty models, it achieved better score with the constant gap model than the affine gap model, but the alignment length increased about 9% for some sequences. By using the EUMed Grid, it was possible to increase the number of used particles and achieve better alignment.
The proposed algorithm also showed good scalability vs. the number of nodes in the cluster and achieved almost 30 times speedup using 6 nodes. It also proved to work well with very long sequences (up to 10000 residues). The main advantages of the proposed parallel PSO algorithm is its ability to explore more search space than the sequential PSO algorithm without affecting the processing time and its support of different objective functions and substitution matrices.
In future research, more tests will be applied on the proposed algorithm using different benchmarks, other substitution matrices will be supported and other mutation techniques will be investigated to improve the initial generated particles.
REFERENCES
- Akioka, S. and Y. Muraoka, 2010. HPC benchmarks on amazon EC2. Proceedings of the IEEE 24th International Conference on Advanced Information Networking and Applications Workshops, April 20-23, 2010, Perth, WA, pp: 1029-1034.
CrossRef - De Almeida, T.J.B.M. and N.F.V. Roma, 2010. A parallel programming framework for Multi-core DNA sequence alignment. Proceedings of International Conference on Complex Intelligent and Software Intensive Systems, February 15-18, 2010, Krakow, pp: 907-912.
CrossRef - Boukerche, A., A.C.M. Alves de Melo, E.F. de Oliveira Sandes and M. Ayala-Rincon, 2007. An exact parallel algorithm to compare very long biological sequences in clusters of workstations. Cluster Comput., 10: 187-202.
CrossRef - Buck, I., T. Foley, D. Horn, J. Sugerman, K. Fatahalian, M. Houston and P. Hanrahan, 2004. Brook for GPUs: Stream computing on graphics hardware. ACM Trans. Graphics, 23: 777-786.
CrossRef - Chamberlain, B.L., S.E. Choi, E.C. Lewis, L. Snyder, W.D. Weathersby and C. Lin, 1998. The case for high level parallel programming in ZPL. IEEE Comput. Sci. Eng., 5: 76-86.
CrossRef - Da Silva, F.J.M., J.M.S. Perez, J.A.G. Pulido and M.A.V. Rodriguez, 2008. AlineaGA: A genetic algorithm for multiple sequence alignment. New Challenges Applied Intelligence Technol., 134: 309-318.
CrossRef - Da Silva, F.J.M., J.M.S. Perez, J.A.G. Pulido and M.A.V. Rodriguez, 2011. A parallel niched pareto evolutionary algorithm for multiple sequence alignment. Practical Appli. Comput. Biol. Bioinform., 193: 157-165.
CrossRef - Dayhoff, M.O., R.M. Schwartz and B.C. Orcutt, 1978. A model of evolutionary change in proteins. Atlas Protein Sequence Structure, 5: 345-352.
Direct Link - Elbeshehy, E.K.F. and A.A.A. Sallam, 2012. Partial characterization of an isolate of cucumber mosaic virus from ismailia governorate. Int. J. Virol., 8: 90-97.
CrossRef - Elkalamawy, I.M., S. Elhddad, M.A. Swelim, S.M. Hamdy and A.H. Fahmy, 2011. Molecular variability of the S gene of hepatitis B virus in Egypt. Int. J. Virol., 7: 109-115.
CrossRef - EI-Kholy, A.A., S. Vilcek and A.M. Daoud, 2005. Phylogenetic characterization of some bovine viral diarrhea viruses in Egypt. Int. J. Virol., 1: 41-41.
CrossRefDirect Link - Nur Fariza, M.S., S.L. Pang, C.Y. Choong and R. Wickneswari, 2008. Extensive DNA sequence variations in two lignin genes, Cinnamate 4-hydroxylase and Cinnamyl alcohol dehydrogenase from Acacia mangium and Acacia auriculiformis. J. Boil. Sci., 8: 687-690.
CrossRefDirect Link - Gropp, W., 2002. MPICH2: A new start for MPI implementations. Lecture Notes Comput., 2474: 37-42.
CrossRef - Hassanain, M.A., F.A.M. Khalil, K.A. AbdEl-Razik and R.M. Shaapan, 2011. Prevalence and molecular discrimination of Cryptosporidium parvum in calves in Behira provinces, Egypt. Res. J. Parasitol., 6: 101-108.
CrossRefDirect Link - Henikoff, S. and J.G. Henikoff, 1992. Amino acid substitution matrices from protein blocks. Proc. Nat. Acad. Sci. USA., 89: 10915-10919.
Direct Link - Jeanmougin, F., J.D. Thompson, M. Gouy, D.G. Higgins and T.J. Gibson, 1998. Multiple sequence alignment with Clustal X. Trends Biochem. Sci., 23: 403-405.
CrossRef - Joo, K., J. Lee, I. Kim, S.J. Lee and J. Lee, 2008. Multiple sequence alignment by conformational space annealing. Biophys. J., 95: 4813-4819.
CrossRef - Kuracha, M.R., B. Rayavarapu, V.R.S.K. Duvvuri and P.N. Rao, 2006. Comparison of secondary structure of the ribosomal internal transcribed spacer 2 of eight Lepidopteran species from diverse geographical locations. J. Entomol., 3: 222-230.
CrossRefDirect Link - Luebke, D., 2008. CUDA: Scalable parallel programming for High-performance scientific computing. Proceedings of the 5th IEEE International Symposium on Biomedical Imaging: From Nano to Mirco, May 14-17, 2008, Paris, pp: 836-838.
CrossRef - Lee, J., J. Kim, S. Seo, S. Kim and J. Park et al., 2010. An OpenCL framework for heterogeneous multicores with local memory. Proceedings of the IEEE 19th International Conference on Parallel Architectures and Compilation Techniques, September 11-15, 2010, Vienna, Austria, pp: 193-204.
CrossRef - Li, K.B., 2003. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics, 19: 1585-1586.
CrossRef - Ligowski, L. and W. Rudnicki, 2009. An efficient implementation of Smith Waterman algorithm on GPU using CUDA, for massively parallel scanning of sequence databases. Proceedings of IEEE International Symposium on Parallel and Distributed Processing, May 23-29, 2009, Rome, pp: 1-8.
CrossRef - Mohsen, M.S. and R. Abdullah, 2011. HS-MSA: New algorithm based on Meta-heuristic harmony search for solving multiple sequence alignment. Int. J. Comput. Sci. Inform. Security, 9: 70-85.
Direct Link - Nordin, M. and A. Rahman, 2009. Utilizing MPJ express software in parallel DNA sequence alignment. Proceedings of the International Conference on Future Computer and Communication, April 3-5, 2009, Kuala Lumpar, pp: 567-571.
CrossRef - Notredame, C., 2002. Recent progresses in multiple sequence alignment: A survey. Pharmacogenomics, 3: 131-144.
PubMed - Opabode, J.T., O.O. Oyelakin , O.A. Akinyemiju and I.L. Ingelbrecht, 2011. Isolation of genomic clones encoding Granule-Bound Starch Synthase (GBSS I) in Cassava (Manihot esculenta Crantz). J. Plant Sci., 6: 174-181.
CrossRefDirect Link - Qasem, J.A., S. Al-Zenki and A. Al-Marzouk, 2010. Identification and characterization of Streptococcus agalactiae Isolates using 16S rRNA sequencing and cellular fatty acid composition analysis. Pak. J. Biol. Sci., 13: 9-15.
CrossRefPubMedDirect Link - Rahim, F., 2010. An in silico development of selective inhibitor for histamine receptors. Biotechnology, 9: 157-163.
CrossRefDirect Link - Salam, R.A., R. Abdullah, M.F. Omar and N.A. Rashid, 2005. Multiple sequence alignment using optimization algorithms. World Acad. Sci. Eng. Technol.
Direct Link - Sarje, A. and S. Aluru, 2008. Parallel biological sequence alignments on the cell broadband engine. Proceedings of IEEE International Symposium on Parallel and Distributed Processing, April 14-18, 2008, Miami, FL, pp: 1-11.
CrossRef - Sarkar, S., T. Majumder, A. Kalyanaraman and P.P. Pande, 2010. Hardware accelerators for biocomputing: A survey. Proceedings of IEEE International Symposium on Circuits and Systems, May 30-June 2, 2010, Paris, pp: 3789-3792.
CrossRef - Suneetha, D.R.S., P.A. Babu and P.V. Joshua, 2008. The phylogenetic analysis of animal and plant D-type cyclins. Trends Bioinform., 1: 25-32.
CrossRefDirect Link - Tambunan, U.S.F., H.W. Butar-Butar, R. Umbas and Z. Hidayah, 2007. Conserved region analysis of oncogenic human Papillomavirus genome. Biotechnology, 6: 93-96.
CrossRefDirect Link - Thompson, J.D., F. Plewniak and O. Poch, 1999. BAliBASE: A benchmark alignments database for the evaluation of multiple sequence alignment programs. Bioiformatics, 15: 87-88.
CrossRef - Van Walle, I., I. Lasters and L. Wyns, 2005. SABmark-a benchmark for sequence alignment that covers the entire known fold space. Bioinformatics, 21: 1267-1268.
CrossRef - Joshua, P.V., D.R. Salomi Suneetha, A. Arundhati and G. Seshagiri Rao, 2010. Studies on Agrobacterium rol gene homologues in Nicotiana rustica L., N. plumbaginifolia viv. and their hybrid. Int. J. Bot., 6: 280-286.
CrossRefDirect Link