
A.C. Kumoro
Department of Chemical Engineering, Faculty of Engineering, Diponegoro University Prof. H. Soedarto, SH Road, Tembalang-Semarang 50239, Indonesia
LiveDNA: 62.4759
D.S. Retnowati
Department of Chemical Engineering, Faculty of Engineering, Diponegoro University Prof. H. Soedarto, SH Road, Tembalang-Semarang 50239, Indonesia
C.S. Budiyati
Department of Chemical Engineering, Faculty of Engineering, Diponegoro University Prof. H. Soedarto, SH Road, Tembalang-Semarang 50239, Indonesia
American Journal of Food Technology
Year: 2010 | Volume: 5 | Issue: 2 | Page No.: 100-110







ABSTRACT
The aim of this study were to observe the possibility of application of microwave heating in the acetylation of cassava starch and to study the physicochemical properties of the starch acetate obtained. The acetylation was carried out by mixing native cassava starch with chloroacetic acid and sodium hydroxide of a certain weight ratio in a sealed container. The mixture was then sprayed with ethanol and heated using microwave power. The Degree of Substitution (DS), Reaction Efficiency (RE) and some physical properties of the acetylated starches were then analyzed. It was found that microwave assisted acetylation of cassava starch using chloroacetic acid can be done in a very short reaction time. The highest DS and RE obtained were 0.045 and 0.051%, respectively. Acetylation of cassava starch reduced gel hardness during storage. Acetylation also inhibits the retrogradation of starch gel. Cassava starch acetylation changed starch molecular motion, resulting in a decrease in the glass transition temperature. Amylopectin retrogradation was not significantly reduced, indicating that the degrees of modification of the starches in this study were too low to cause enough steric hindrance to prevent retrogradation. The modifications were done on native starch granules; they took place preferentially on the amylose fraction, not the amylopectin fraction, thus leaving amylopectin retrogradation was mostly unaffected. It can be concluded that microwave heating can be applied in the acetylation of cassava starch to obtain significant changes of the properties of starch.
PDF Abstract XML References Citation
How to cite this article
A.C. Kumoro, D.S. Retnowati and C.S. Budiyati, 2010. Microwave Assisted Synthesis and Characterization of Acetate Derivative Cassava Starch. American Journal of Food Technology, 5: 100-110.
DOI: 10.3923/ajft.2010.100.110
URL: https://scialert.net/abstract/?doi=ajft.2010.100.110
DOI: 10.3923/ajft.2010.100.110
URL: https://scialert.net/abstract/?doi=ajft.2010.100.110
INTRODUCTION
Indonesia is one of the world’s largest cassava producers and cassava has become the most economic source of starch in the country. Cassava starch is appreciated for its paste clarity, low gelatinization temperature, good gel stability and low tendency to syneresis (Sedas and Kubiak, 1994). However, it has disadvantages that would make it unsuitable for food systems and processing such as: narrow peak viscosity range, undesirable texture, poor stability and processing tolerance (Mali and Grossmann, 2001). Modification of native starches is thSerefore, necessary to improve their desirable functional properties (Han et al.,2005). Modified starches have wide applications in industries, such as thickeners, binders, fillers, emulsion stabilizers, consistency modifiers and adhesives (Guilbot and Mercier, 1985; Ogura, 2004).
Starch modification methods can be classified into four categories, namely: physical, chemical, enzymic and biological modifications (BeMiller, 1997; Yiu et al., 2008). However, chemical modification is the most frequently used process due to its superiorities compared to other aforementioned methods (John and Raja, 1999). Acetylation of cassava starch through the introduction of acetyl group is expected to reduce the interaction between starch molecules and thereby increase the swelling power (Rutenberg and Solarek, 1984) and water solubility of the starch granules (Aziz et al., 2004). Starch acetates and other esters can be made very efficiently on a micro scale without addition of catalyst or water simply by heating dry starch with acetic acid and anhydride at 180°C for 2-10 min (Shogren, 2003). At this temperature, starch will melt in acetic acid (Shogren, 2000) and thus, a homogeneous acetylation would be expected to occur. In order to scale up this process, microwave heating was chosen since it gives fast, uniform heating and sometimes enhanced reaction rates. Recently, microwave techniques find application in many diverse areas of chemistry, including analytical chemistry, organic and inorganic synthesis, manufacturing of ceramics, pharmaceutical chemistry and catalysis (Mingos and Baghurst, 1991; England, 2003; Shogren, 2003). Koroskenyi and McCarthy (2002) found that microwave heating accelerated the esterification and etherification of starches. During ultrafast synthesis using microwave irradiation, reactions are completed in minutes compared to hours or days using the conventional methods. Systematic studies on microwave-aided chemical modifications of starch are scarce and no comprehensive literature seems to exist on the use of microwaves for the acetylation of starch.
The objective of this study has been to employ microwave heating for the synthesis of acetate derivatives of cassava starch using chloroacetic acid and to study the physicochemical properties of the products.
MATERIALS AND METHODS
Materials
Native cassava starch used in this research was purchased from local market nearby Diponegoro University campus in the mid of June 2009 when this research project was just started. Aquadest was supplied from Reverse Osmosis Unit available in the Department of Chemical Engineering, Faculty of Engineering, Diponegoro University. In addition, sodium hydroxide, chloroacetic acid and all reagents used for analysis were purchased from local authorized distributor for Sigma-Aldrich Co. Inc. of their Analytical Grade.
Equipment
The main equipment used in this work comprises microwave oven, integrated thermo controller and indicator, rapid visco analyzer, texture analyzer, temperature controlled water bath, agitator and its electrical motor, suction pump, centrifuge and glass wares. Digital Scanning Calorimetry (DSC) analyzer to investigate the thermal properties of starch was partly used in the Integrated Analytical Laboratory Unit, Gadjah Mada University, Yogyakarta.
Procedures
Preparation of Acetylated Cassava Starch in a Batch System
Modification of native cassava starch was done by acetylation method assisted by microwave heating as previously mentioned by Zhongdong (1998) with slight modification. Native cassava starch (16.8 g), sodium hydroxide (8.8 g) and chloroacetic acid (8.5 g) were mixed thoroughly in a 300 mL Teflon vessel. A magnetic stir bar was added and the mixture was stirred for 5 min. Ethanol of 50 mL was then sprayed into the mixture using spry catcher. The vessel was then sealed, the thermocouple inserted and the vessel was heated in a microwave labstation 1600 (Milestone, Inc., Shelton, CT). Then, the mixture was subjected to microwaving (hold stability, without moving), for 2-10 min. Power to the microwave magnetron was automatically adjusted to give the linear temperature ramps entered into the computer control system. Maximum microwave power was limited to 650 W to avoid overshooting the temperature program. Unless otherwise stated, the temperature ramp was 25-150°C over 3.5 min, 150-160°C over 1.5-4.5 min. The acetylated cassava starch was then ready for analysis.
Analysis
Starch Proximate Analysis
Proximate composition of samples was determined according to the methods of Association of Official Analytical Chemists (AOAC, 1980) with analytical codex number 14.062, 14.064, 14.066 and 14.067 for moisture, total ash, total crude fibre, crude fat and total crude protein, respectively. Carbohydrate is obtained by difference. All results are the average of duplicate analyses.
Determinations of Degree of Substitution
Acetyl contents of Acetylated Starch (AS) were determined using the method of Wurzburg (1964) with a slight modification. Starch (1 g) was suspended in 20 mL of a 78% ethanol solution. The slurry was kept in a water bath at 50°C for 30 min. Then the slurry was cooled and 30 mL of 0.5 M potassium hydroxide was added. After stirring for 72 h at room temperature, the excess of alkali was titrated with 0.5 M hydrochloric acid using phenolphthalein as an indicator. A blank was titrated at the same time. Acetyl content (A) was calculated using the equation:
| (1) |
where V0 is mL of 0.5 M HCl used to titrate the blank; Vn is mL of 0.5 M HCl used to titrate the sample; M is molarity of used HCl; W is weight of sample used (g) and 43 is the molecular weight of the acetyl group.
The degree of substitution in AS and ACS was as follows:
| (2) |
where, A is the acetyl content of modified starches.
Reaction efficiencies were based on the amount of chloroacetic acid added and were calculated as:
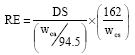 | (3) |
where, wca and wcs are the weights of chloroacetic acid and starch, respectively.
Pasting Behavior
Pasting characteristics of the native starch and citrates were studied using a Rapid Visco Analyzer (RVA- 4, Newport Scientific, Warriewood, NSW, Australia). The slurries (100 g kg-1 by weight) were heated from 50 to 95°C at 12°C min-1 and these were then held at 95°C for 2 min. The pastes were cooled to 50°C at 12°C min-1 and finally kept at 50°C for 2 min. The values recorded were: peak viscosity (maximum viscosity attained by the starch paste); holding or hot paste viscosity (hold), i.e., minimum viscosity during stirring at 95°C; breakdown viscosity (peak-hold); final viscosity, i.e., viscosity at the end of the cycle at 50°C; setback viscosity (final-hold); and pasting temperature. The viscosity analysis was done in duplicate.
Thermal Characteristic
The thermal properties of selected samples were determined using a Mettler Toledo differential scanning calorimeter (Mettler, Schcoerfenbach, Switzerland). Sample (5 mg) was weighed into a pre-weighed aluminium pan and 10 μL distilled water was added. The pan was sealed hermetically and transferred to the heating chamber of the calorimeter. An empty pan was used as reference and the sample was heated from 20 to 100°C at a rate 10°C min-1 and cooled back to 20°C at the same rate. The temperatures corresponding to the onset of gelatinization (To), peak (Tp) and endset (Te) and also the heat of gelatinization (ΔH) were recorded. Samples were analyzed in duplicates and the data presented are the average of the two measurements.
Gel and Retrogradation Characteristic
Gel Preparation
For each starch sample, starch and distilled water were mixed in the proportion 1:1 by weight. Before hand the distilled water was boiled and cooled down to room temperature in a hermetically sealed Erlenmeyer flask in order to remove dissolved gases and reduce the formation of bubbles in the samples. The resulting slurry was transferred into polypropylene tubes 9 cm long and 2 cm wide. The tubes were heat sealed and progressively heated in a water bath to 60, 70 and finally 95°C, in order to obtain starch gels with homogenous hydration. The total gelatinization time was 80 min. The samples were then transferred onto aluminum trays and left to cool down to room temperature (257°C) for 30 min. On the day of preparation (day 0), the texture of each gel was measured. The moisture contents of the gels were determined in triplicate by drying 3.5 g of gels at 105°C for 18 h.
Gel Texture Analysis
The gel samples had a cylindrical shape approximately 60 mm long. The central part of the gel was cut for texture analysis to the following dimensions: 30 mmlongx610 mm diameter. The analysis was done with a Stable Microsystems TA-XTPlus (Godalming, Surrey, UK) fitted with a 30 kg load cell and a 5 cm-diameter aluminum cylindrical probe. The conditions of the test were adapted from the method suggested for a single compression test in the texture analyzer manual. The gels were laid out horizontally on the texture analyzer platform. A single compression protocol was chosen, with the target compression set to 40% of the initial height, corresponding to a compressed height of 3 to 4 mm depending on the exact initial height. The probe speed was set to 1 mm sec-1. The gel initial height was measured automatically at the beginning of the measurement, when the probe first touched the gel surface, using a contact force for sample detection of 0.1 g. Because some gels shattered when compressed more than 3 mm, the parameter force at 2 mm compression was chosen as representative of the hardness of the gels, in order to be able to compare all the samples. Measurements were done in triplicates for all samples.
Gel Storage
The samples not used on day 0 were stored for eight days in a refrigerator at 4°C. On days 1, 3 and 7, three tubes of each sample were taken for texture and DSC analysis. Because the aim of this study was to study amylopectin retrogradation, the storage conditions were chosen to be close to the maximum retrogradation rate, as predicted by the Lauritzen-Hoffman model of polymer crystallization (Farhat et al., 2000), gel moisture content approximately 50% wwb and storage temperature 4°C. Also, the storage time was chosen so as to match the timescale for amylopectin retrogradation. The retrogradation of amylose, which occurs over a few hours after cooling post-gelatinization (Fechner et al., 2005) was not monitored.
Statistical Analysis
The design was analyzed using the statistical package SAS 8.01 to fit the polynomial equations to data and to generate response surface plots using significant parameters (p<0.05) (Xie and Liu, 2004).
RESULTS
Proximate Composition
Proximate composition of the native cassava starch used in this research was: fat, 0.06%; protein, 0.34%; ash, 0.50%; moisture, 16.00%; and carbohydrate, 95%. As can be seen in Table 1, these findings are in agreement with the earlier report of Charles et al. (2005) and Sriroth et al. (2000). The moisture content of starches varied from 9.6 to 16%, placing the technological classification of this product as a moderately hygroscopic product.
Effect of Reaction Time on the Degree of Substitution of Modified Cassava Starch
As can be seen in Fig. 1, the highest DS value of 0.045 represents 0.051% reaction efficiency was achieved within 6 min of reaction time. Longer reaction times were observed by Mark and Mehhltretter (1972), Billmers and Tessler (1993) and Shogren (1996). For all experiments, reaction temperatures were increased linearly from 25 to 150°C over 3.5 min then increased linearly from 150-160°C over the remainder of the time indicated.
| Table 1: | Proximate analysis of native cassava starch |
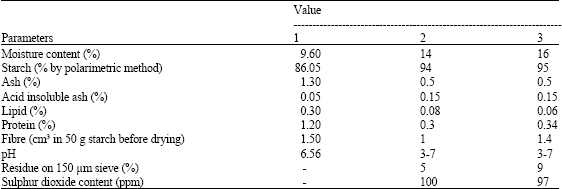 | |
| 1: Charles et al. (2005); 2: Sriroth et al. (2000); 3: Present study | |
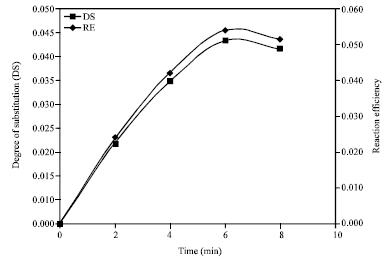 |
| Fig. 1: | Effect of microwave heating time on the degree of substitution and reaction efficiency of cassava starches acetylation |
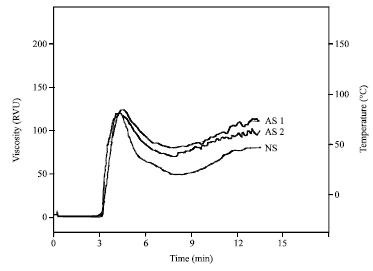 |
| Fig. 2: | RVA pasting profile of acetylated (AS1 and AS2) and native cassava starches |
Viscosity Characteristic of Modified Cassava Starch
From Fig. 2, it is obvious that the acetylated starches (AS1 and AS2) have similar characteristics to the non-modified starch in terms of peak viscosity (340-360 RVU) and setback viscosity (85.0-96 RVU). Low degree acetylated cassava starch (DS = 0.020) has higher pasting temperature and higher final viscosity compared to high degree acetylated cassava starch (DS = 0.045). It can also be seen in Table 2, that acetylation of cassava starch lead to increase the resistance of the starch granules to shear.
Thermal Properties of Modified Cassava Starch
The moisture content of the starches investigated in this work were within 12.0-12, 7.5% weight wet basis (wwb), with standard deviation of 0.02-0.20. From Table 3, it is observed that native cassava starch possesses highest glass transition temperature, which is 91.4°C. On the other hand, high degree substitution acetylated starch (DS = 0.045) has lowest glass transition temperature (83.9°C).
| Table 2: | Pasting properties of acetylated (AS1 and AS2) and native cassava starches |
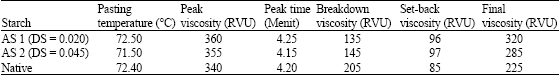 | |
| Table 3: | Glass transition temperature and variation in specific heat for native and acetylated cassava starches at 12% moisture content (wet basis) |
 | |
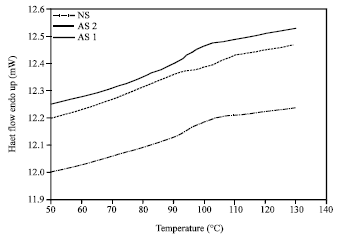 |
| Fig. 3: | The DSC thermogram acetylated (AS1 and AS2) and native cassava starches containing 12% moisture |
Low degree substitution acetylated starch (DS = 0.020) has slightly higher glass transition temperature (85.60°C). It is also obvious in Fig. 3 that acetylation of cassava starch reduced the glass transition temperature. Table 3 also shows that modification of cassava starch through acetylation had little effect on the specific heat of the glass transition, with no significant differences between two acetylated starch samples with the native starch.
Gel Properties of Modified Cassava Starch
Fresh gel obtained from native and acetylated starch were clear, translucentid and sticky. Aged gels had taken a creamy white color, indicative of the formation of starch crystallites during retrogradation. No stickiness was found in the aged gels. Table 4 shows that compared to the non-modified starch fresh gel hardness (171.1 g), acetylation at DS 0.020 and 0.045 levels significantly reduced fresh gel hardness (96.5 and 105.5 g for the AS1 and AS2 samples, respectively).
As can be seen in Table 5, the hardness of all starch gels increased in the first three days, before reaching a plateau of maximum hardness during storage. Experimental result shows that native cassava starch and low degree substitution acetylated starch were categorized as intermediate gel hardness starch. While, higher degree substitution acetylated starch was classified as low gel hardness starch.
| Table 4: | Hardness (force at 2 mm compression, in grams) of fresh gels made from native and acetylated cassava starches |
 | |
| Table 5: | Hardness (force at 2 mm compression, in grams) of gels made from native and acetylated starches during seven days of storage |
 | |
| Table 6: | Retrogradation enthalpies of native and acetylated cassava starch gels after seven days of storage (J g-1) |
 | |
Retrogradation of Modified Cassava Starch
Table 6 shows that retrogradation enthalpy of cassava starch gel increased with degree of substitution (DS = 0.045) for seven days. Moisture contents of the observed gel were between 52-55% weight wet basis. In the fresh gels (day 0), no retrogradation was detected. Retrogradation enthalpy of amylopectine of acetylated starch increased from day 1 to 3. High value of retrogradation enthalpy indicated that the starch is not prone to retrogradation. While retrogradation enthalpy of native cassava starch decreased from day 1 to day 3 and increased back approaching its day 1 retrogradation enthalpy after seven days of storage.
DISCUSSION
The differences between the proximate composition of the starch obtained in this study and the reported data were possibly caused by different cultivar of the cassava, treatments and processing conditions. The native cassava starch used in this work was made traditionally by home industries nearby Diponegoro University campus.
The difference in reaction time consumed in this work with the previous works was mainly caused by different acetic source used, temperature, homogeneity of the reaction media and heating mode. Shogren (1996) added NaOH as catalyst into cool starch and acetic anhydride to obtain starch acetate of DS 2.5 in 50 min, while Mark and Mehltretter (1972) preheated starch and acetic anhydride before adding NaOH to obtain the same DS in about 3 h. The starch may have had more time to be swollen by water and NaOH when heating was gradual. Billmers and Tessler (1993) prepared DS 0.5-l-8 granular starch esters by reaction of starch with anhydrides and NaOH in aqueous suspension. Reaction times were even longer (2-8 h), possibly because of the need to hold reaction temperatures at lower than 40°C to avoid gelatinization and dilution of the reaction with large amounts of water.
The decreasing rate of reaction with increasing amylose content may be due to the presence of crystalline amylose or amylose complexes. These crystalline forms normally melt at a higher temperature than the native crystalline amylopectin component (Shogren, 1992) and thus, would be more resistant to swelling and reaction. Shogren (2000) also explained that polysaccharides are much more reactive after first swelling and, particularly, solubilization or melting in a suitable solvent. Crystalline form and highly hydrogen-bonded domains react slowly since, access of the reagent is poor and mobility is low. In addition, the decrease in acetylation rate might also be influenced by the increase of degree of substitution of the starch (Shogren, 1996).
The acetylation procedure apparently caused little reduction in the molecular weight of the starch and increased acetylated cassava starch granule resistance to shear as indicated by lower breakdown viscosity of acetylated cassava starch (135-145 RVU) compared to that of native cassava starch (205 RVU). For the case of starch esters made by the homogeneous microwave reaction, there is possibly a more random distribution of constituents over the whole starch molecule thus opening up the conformation to interaction with water and other hydrophobes. Intrinsic viscosities and hence, molecular weights are much lower for the microwave synthesized starch esters than those made by aqueous suspension reaction (Shogren and Biresaw, 2007). This is in agreement with the data of Mark and Mehltretter (1972) for DS 3 starch acetates and Shogren (1996).
As mentioned by polymer theory side functional groups contained in the polymer structure may play two opposite roles. They can either increase the stiffness of the polymer backbone and increase the glass transition temperature, or increase the free volume between polymer chains and thereby reduce the glass transition temperature. In this study, the effect of acetylation was on the side of an increase in free volume and molecular mobility. Replacement of hydroxyl group by acetyl groups possibly decreased the extent of intermolecular hydrogen bonding and allowed more molecular mobility in the dry state (Shogren, 1996). Therefore, the glass transition temperatures of acetylated starches were found lower than that of native cassava starches.
The findings obtained in this work indicated that the degree of acetylation was the main factor determining the texture of starch gels during storage even at low degrees of substitution. Acetylation highly improved gel stability of the paste from native starch granules (Hung and Morita, 2005). Gel stability of the acetylated starches was significantly higher than that of native starch with significantly lower water released from the fresh paste and after freeze-thaw. It is likely that the underlying molecular mechanism is steric hindrance limiting the formation of starch double helixes. However, this finding was in contradiction with acetylation of cocoyam, which retrogradation increased with degree of substitution.
CONCLUSIONS
Microwave assisted acetylation of cassava starch can be done in a just 6 min. Modification of the starch caused significant changes to the properties of starch. Acetylation of cassava starch reduced gel hardness during storage. Acetylation also inhibits the retrogradation of starch gel. Cassava starch acetylation changed starch molecular motion, resulting in a decrease in the glass transition temperature. Amylopectin retrogradation was not significantly reduced, indicating that the degrees of modification of the starches in this study were too low to cause enough steric hindrance to prevent retrogradation. The modifications were done on native starch granules; they took place preferentially on the amylose fraction, not the amylopectin fraction, thus leaving amylopectin retrogradation mostly unaffected.
ACKNOWLEDGMENT
The researchers would like to express their gratitude to the Directorate General of Higher Education, Ministry of National Education for its financial support through Competitive National Priority Research Grant 2009 through contract agreement No. 304/SP2H/PP/DP2M/VI/2009.
REFERENCES
- AOAC., 1980. Official Methods of Analysis. 13th Edn., Association of Official Analytical Chemist, Washington, DC., USA., pp: 56-132.
Direct Link - Aziz, A., R. Daik, M.A. Ghani, N.I.N. Daud and B.M. Yamin, 2004. Hydroxypropylation and acetylation of sago starch. Malaysian J. Chem., 6: 48-54.
Direct Link - BeMiller, J.N., 1997. Starch modification: Challenges and prospects. Starch Starke, 49: 127-131.
CrossRef - Charles, A.L., K. Sriroth and T.C. Huang, 2005. Proximate composition, mineral contents, hydrogen cyanide and phytic acid of 5 cassava genotypes. Food Chem., 92: 615-620.
CrossRefDirect Link - Farhat, I.A., J.M.V. Blanshard and J.R. Mitchell, 2000. The retrogradation of waxy maize starch extrudates: Effects of storage temperature and water content. Biopolym., 53: 411-422.
CrossRef - Fechner, P.M., S. Wartewig, P. Kleinebudde and R.H.H. Neubert, 2005. Studies of the retrogradation process for various starch gels using Raman spectroscopy. Carbohydr. Res., 340: 2563-2568.
CrossRef - Han, J.A., B.H. Lee, W.J. Lim and S.T. Lim, 2005. Utilization of hydroxypropylated waxy rice and corn starch in Korean waxy rice cake to retard retrogradation. Cereal Chem., 82: 88-92.
CrossRef - Hung, P.V. and N. Morita, 2005. Effects of granule sizes on physicochemical properties of cross-linked and acetylated wheat starches. Starch, 57: 413-420.
CrossRefDirect Link - John, J.K. and K.C.M. Raja, 1999. Properties of cassava starch-dicarboxylic acid complexes. Carbohydr. Polym., 39: 181-186.
CrossRef - Koroskenyi, B. and S.P. McCarthy, 2002. Microwave-assisted solvent-free or aqueous-based synthesis of biodegradable polymers. J. Polym. Environ., 10: 93-104.
CrossRef - Mali, S. and M.V.E. Grossmann, 2001. Preparation of acetylated distarch adipates by extrusion. Lebensmittel-Wissenschaft und-Technol., 34: 384-389.
CrossRef - Mark, A.M. and C.L. Mehltretter, 1972. Facile preparation of starch triacetates. Starch, 24: 73-76.
CrossRef - Mingos, D.M.P. and D.R. Baghurst, 1991. Application of microwave dielectric heating effects to synthetic problems in chemistry. Chem. Soc. Rev., 20: 1-47.
CrossRefDirect Link - Shogren, R.L., 1992. Effect of moisture content on the melting and subsequent physical aging of cornstarch. Carbohydr. Polym., 19: 83-90.
CrossRef - Shogren, R.L., 1996. Preparation, thermal properties, and extrusion of high-amylose starch acetates. Carbohydr. Polym., 29: 57-62.
CrossRef - Shogren, R.L., 2000. Modification of maize starch by thermal processing in glacial acetic acid. Carbohydr. Polym., 43: 309-315.
CrossRef - Shogren, R.L., 2003. Rapid preparation of starch esters by high temperature/pressure reaction. Carbohydr. Polym., 52: 319-326.
CrossRef - Shogren, R.L. and G. Biresaw, 2007. Surface properties of water soluble maltodextrin, starch acetates and starch acetates/alkenylsuccinates. Colloids Surfaces A: Physicochem. Eng. Aspects, 298: 170-176.
CrossRef - Sriroth, K., K. Piyachomkwan, S. Wanlapatit and C.G. Oates, 2000. Cassava starch technology: The Thai experience. Starch, 52: 439-449.
CrossRefDirect Link - Wurzburg, O.B., 1964. Starch Derivatives and Modification Acetylation. In: Methods in Carbohydrate Chemistry, Whistler, R.L., E.F. Paschall (Eds.). Vol. 4, Academic Press, New York, ISBN: 0127462031, pp: 335.
Direct Link - Xie, X. and Q. Liu, 2004. Development and physicochemical characterization of new resistant citrate starch from different corn starches. Starch, 56: 364-370.
CrossRef - Yiu, P.H., S.L. Loh, A. Rajan, S.C. Wong and C.F.J. Bong, 2008. Physiochemical properties of sago starch modified by acid treatment in alcohol. Am. J. Applied Sci., 5: 307-311.
Direct Link - Zhongdong, L., 1998. Preparation of wheat carboxymethyl starch (CMS) under microwave and the structural analysis. Chem. Reactn. Eng. Technol., 14: 243-250.
Direct Link