
P.T.V. Lakshmi
Department of Bioinformatics, Phytomatics Laboratory, Bharathiar University, Coimbatore-46, Tamil Nadu, India
S. Radhika
Department of Bioinformatics, Phytomatics Laboratory, Bharathiar University, Coimbatore-46, Tamil Nadu, India
A. Annamalai
Department of Biotechnology, Plant Cell and Molecular Biology Laboratory, Karunya University, Coimbatore, Tamil Nadu, India
Trends in Bioinformatics
Year: 2011 | Volume: 4 | Issue: 1 | Page No.: 23-34







ABSTRACT
The mechanism of hydrolysing the β-lactam antibiotics by β-lactamases is referred to as antibiotic resistance and this could be reduced by introducing some β-lactamase inhibitors. However, surprisingly these inhibitors also have the property to defend bacteria and therefore are used in combination with antibiotics which still gain resistance owing to the property of multi-drug resistance by the use of MRSA (Methicillin-resistant Staphylococcus aureus). Therefore, the present study was focused on the inhibitory activity of a set of natural compounds (Gallic acid, Propyl Gallate, Kaempferol, Quercetin, Mangostin and Rubraxanthone) from natural resources against β-lactamase in MRSA by insilico interaction studies using Autodock4 suite. Their corresponding interactions at the binding sites were studied after inducing the solvation properties. Before performing the hydrogen bond interactions the ligands were subjected to ADMEtoxicity (Absorption, distribution, Metabolism and Excretion) analysis and were further examined to find out the optimum pose of the ligands in order to choose the best among them. However, Kaempferol exhibited best interactions with minimum inhibitory constant and thus was selected as the best inhibitor for this study.
PDF Abstract XML References Citation
Received: March 14, 2011;
Accepted: May 28, 2011;
Published: June 29, 2011
How to cite this article
P.T.V. Lakshmi, S. Radhika and A. Annamalai, 2011. Molecular Docking Analysis of Phyto-Ligands with Multi Drug Resistant β-lactamases of Staphylococcus aureus. Trends in Bioinformatics, 4: 23-34.
DOI: 10.3923/tb.2011.23.34
URL: https://scialert.net/abstract/?doi=tb.2011.23.34
DOI: 10.3923/tb.2011.23.34
URL: https://scialert.net/abstract/?doi=tb.2011.23.34
INTRODUCTION
“Antibiotic resistance” is the ability of the microorganism to multiply continuously in the presence of antibiotic agents. β-lactam antibiotics such as penicillins, cephalosporins, cephamycins, ertapenems and carbapenems have been recognized to have the capability of destroying the activity by hydrolyzing the lactam ring to confer resistance of an organism against the antibiotics (Bush, 1989). Thus, β-lactam antibiotics are potent, broad-spectrum bactericidal agents with low toxicity to eukaryotes and have widespread clinical uses (Georgopapadakou, 1993). Several resistance mechanisms have been developed in bacteria which include physical removal of antibiotics from the body and modification of target sites (Li et al., 1994). Multi-Drug Resistant Organism (MDRO) viz., Staphylococcus aureus is also resistant to many antibiotics and this resistance perhaps, makes certain drugs to fail at the time of treatment (McLaughlin et al., 1981; Gold and Moellering, 1996). For instance, the acquisition of Methicillin resistance has made S. aureus with a system that has made all members of the largest and most constructive family of antimicrobial agents to supersede the β-lactam antibiotics as the remedial agents against these bacteria (Crisostomo et al., 2001).
Over the past two decades, researches are being focussed to improve the structure or design an agent that prevents the degradation by β-lactamases or that could inhibit them irreversibly. Perhaps, such an agent need not possess antibacterial activity of its own but could be used in conjunction with existing β-lactam. However, the potency of the β-lactam/β-lactamase inhibitor combination depends on a number of factors. The efficiency of inhibition can be measured by studying three factors called IC50 of the inhibitor affinity, the induction potential of the inhibitor and the turnover number (Rotschafer and Ostergaard, 1995).
Detailed studies for better understanding of the interactions between β-lactam antibiotics, β-lactamases and β-lactamase inhibitors at molecular level had been conducted to identify the combinations of antibiotic/inhibitor soon after the discovery of clavulanic acid (Sandanayaka and Prashad, 2002). However a massive amount of plant compounds is readily accessible from herbal suppliers and natural-food stores (Aqil et al., 2005). Infect, highly oxidized phenols have been reported to exhibit inhibitory actions on enzymes including, ethanolic extracts. Medicinal plants of India also inhibited beta-lactamase producing Methicillin-Resistant Staphylococcus aureus (MRSA) and Methicillin-Sensitive S. aureus (MSSA). In vitro studies of 10 antibiotics and 15 natural polyphenols against methicillin resistant Staphylococcus aureus revealed quercetin and kaempferol of Laurus nobilis (Lauraceae) among all other polyphenols to exhibit the lowest minimum inhibitory concentrations (Lawson et al., 1989).
The crystal structure of a class-A beta-lacta mase from Staphylococcus aureus PC1 with refinement of 2.0 Å with accession number 3 BLM from RCSB PDB (Herzberg, 1991) was chosen as a target receptor molecule for the present investigation. It consisted of two closely associated domains, each formed by a five-stranded antiparallel beta-sheet with three helices packed against the face of each sheet. The active site was identified to locate in the interface between the two domains and many of the residues that formed it were conserved in all known sequences of class-A beta-lactamases. Another β-lactamase from the same organism but which was crystallized in complex with classical inhibitor clavulanic acid was accessed from RCSB protein data bank using the id 1BLC, wherein, the structure was resolved at 2.2 Å (Chen and Herzberg, 1992). The active site consisted of amino acid residues that bound with inhibitor clavulanic acid by Tyr105, Ser130, Ser216, Ser70, Asn170, Gln237 and Ser235, however, Ser70 was found to be conserved in the active site. This had a structure of two acylenzymes with covalent bonds at the active site Ser70, representing two different stages of inhibitor degradation that alternates the active site (Padayatti et al., 2005).
Usually docking programs are mainly used to discover novel ligands and different databases were screened to check the availability of distinct molecules that efficiently bind with the binding sites on the receptor. Precise prediction of the ligand-receptor complexes is of fundamental importance in modern structure-based drug design (Taylor et al., 2002). Moreover, the ligand matching and their corresponding scoring (Gohlke and Klebe, 2001) could confirm the binding efficiency of each molecule at the binding site, they were evaluated and relatively ranked.
Prediction of ADME properties and toxicity of ligands plays a major role in drug discovery because it mainly aims at finding out disposition behavior of compounds in the whole body by assembling all kinetic properties (Ekins et al., 2000). In silico modeling of ADME properties have been performed using different approaches. AutoDock Tool was employed to study the affinity and binding of a group of plant compounds in the active site of β-lactamase of Staphylococcus aureus. The plant compounds such as Gallic acid, Propyl gallate, Kaempferol, Quercetin, Rubraxanthone and Mangostin chosen were subjected for energy optimization by adding solvation effects and were used further for predicting the binding affinity of the molecules with the receptor proteins. The binding affinity of the ligands with both 1BLC and 3BLM which are the crystallized structures of the same enzyme, were compared. Even though it is very difficult to predict the absorption, distribution, metabolism, excretion and toxicity properties of the ligands (Krejsa et al., 2003), they were determined for the chosen ligands and were further analysed for docking. Docking the protein with the optimized structures of the plant compounds exhibited different poses with minimum energy levels. The most favorable conformation was predicted along with the hydrogen bonding and inhibitory constants which provided the information on the efficiency of the plant inhibitors.
MATERIALS AND METHODS
In the present study, the structure which contained only the enzyme β-lactamase (3BLM) and that existed as a complex with the classical inhibitor clavulanic acid (1BLC) were chosen as targets and a method to inhibit their deleterious activities with the help of ligands from natural resources were tried. The three dimensional structures of β-lactamase from Staphylococcus aureus were retrieved from RCSB PDB site using the PDB ids 3BLM and 1BLC. These structures were opened in Pymol viewer to visualize the active site residues. PDBsum predicted secondary structure of the protein along with the amino acid residues with which the enzyme made up was explained by the secondary pattern of the enzyme, wherein helix and beta sheets were distinguished by the notations H and β. The amino acids which are present in the active site of the enzyme were represented as a dot above the residues which were absent in the x-ray structure of 3BLM. Amino acids from 31 to 290 were shown in the secondary structures of both 3BLM and 1BLC.
The ligands Gallic acid, Propyl gallate, Kaempferol, Quercetin, Mangostin and Rubraxanthone were accessed from NCBI PubChem Compound and were subjected to energy optimization using the quantum chemical package GAMESS. Ab initio quantum chemical calculations which performs Restricted Hartree Fock (RHF) algorithm to calculate the atomic orbital was used to bring the molecules to its minimum energy state. The basis set of the RHF calculation was 6/31-G**. B3LYP had been chosen for the DFT type calculation. ADME properties of the plant inhibitors used for the present study were predicted using ADMETox filtering rules such as molecular weight, polar surface area, logP or number of rotatable bonds from FAFDrugs online server. Docking calculation in AutoDock was performed using the receptor protein and the plant inhibitors and the results were viewed by reading the docking log file (dlg) file in Analyze menu. The dlg file was opened and different conformations were loaded with increasing order of energy. Play option was selected to visualize the conformations. Similarly docking calculation was set for 3BLM with all six ligands and the best binding modes were analyzed.
RESULTS
X-ray crystal structures of β-lactamase from Staphylococcus aureus was viewed in the molecular visualization tool-Pymol. 3BLM had single protein structure whereas 1BLC was a complex molecule with inhibitor clavulanic acid (Fig. 1 a, b). The active sites of 1BLC and 3BLM were obtained from PDBsum of EBI Server which showed to exhibit the secondary structure, displaying the positions of beta turns and alpha helix throughout the entire protein (Fig. 2a, b).
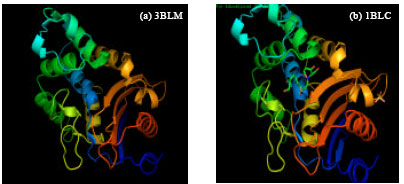 | |
| Fig. 1 (a-b): | X-ray crystal structures of β-lactamase of Staphylococcus aureus retrieved from RCSB PDB |
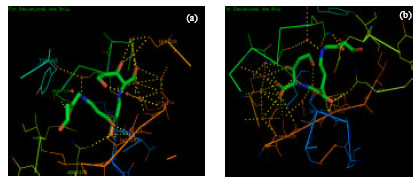 | |
| Fig. 2 (a-b): | Active site views of 1BLC in Pymol. Different poses of interactions involved in the active site of 1BLC are illustrated in (a) and (b). Yellow dotted lines indicate the presence of hydrogen bonds and red dots indicate the presence of water molecules |
The active site predicted by PDBsum was in good agreement with the structure viewed by Pymol. Interestingly, the active site residues found in both structures were found to be highly conserved with the combination of Ala69, Ser70, Lys73, Tyr105, Ser130, Asn132, Glu166, Asn170, Ser216, Lys234, Ser235, GLy236, Gln237, Ile239 and Lys 277. Among these residues Ser70, Lys73, Ser130 and Glu166 were identified as catalytic residues. The conserved pattern of active site residue S-X-X-K was observed in both enzymes chosen, as it normally expressed in all Serine-beta lactamases and penicillin binding proteins (Fan et al., 2007). Ser 70 is followed by threonine and serine at 71st and 72nd position, respectively. The active site thus predicted by PDBsum was viewed efficiently in Pymol and the hydrogen bond interactions were also noted. In the crystal structure of 1BLC the ligand clavulanic acid was found to be efficiently bound to the catalytic as well as active residues. Both covalent and non-covalent interactions were identified. Among non-covalent interactions, hydrogen bond interactions were the most important ones. Water molecules present in the crystal structure had high contribution of hydrogen bonds in the active structure.
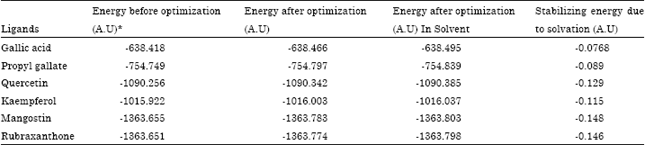
The ligands chosen for the present study were Gallic Acid (GA), Propyl Gallate (PG), Kaempferol (KF), Quercetin (QR), Mangostin (MG) and Rubraxanthon (RX). Ab initio calculations run by GAMESS gave optimized structures of the ligands. The calculations were run under both atmospheric condition and by providing solvation effect by forming a cavity of shell with water. Energy optimization was done for each of the ligands individually with and without solvation. Both the kinetic and potential energies obtained were summed up to produce the final energies as optimized values. The difference between the energy before optimization and after optimization in solvent phase was found to be the factor responsible for the stability and therefore was termed as stabilizing energy in salvation. It revealed the ligands Mangostin, Rubraxanthon, Quercetin and Kaempferol to have greater stabilized energies in the order of -0.148 A.U, -0.146 A.U, -0.129 A.U and -0.115 A.U, respectively (Table 1).
The ligands which were chosen as drug candidates were subjected for predicting the physical and chemical properties using the online tool ADMETox. The predicted features of the ligands revealed Quercetin to contain the highest donor and acceptor atoms, respectively along with larger polar surface area of 132.68. Next larger was revealed by Kaempferol which formed the polar surface area of 107.22 (Table 2).
The ADMETox properties checked and energy optimized ligands of natural origin were subjected to docking analysis to check for its efficiency as an antagonist of β-lactamase. Ligands were docked with the candidate protein by forming grid and saved in docking log file (DLG) format in Autodock4.
| Table 1: | Energy optimization of the Ligands before and after solvation |
 | |
| *The energies calculated for both atmospheric and solvent medium are represented in Atomic Units | |
| Table 2: | ADMETox properties of the ligands |
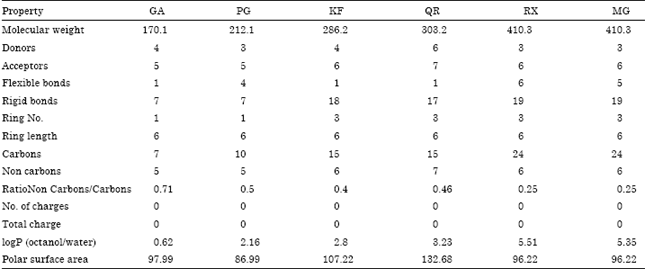 | |
| The double letter coding represents the names of the ligands as: GA: Gallic acid, PG: Propyl gallate, KF: Kaempferol, QR: Quercetin, RX: Rubraxanthone, MG: Mangostin | |
| Table 3: | Docking performance of the ligands with 3BLM and 1BLC |
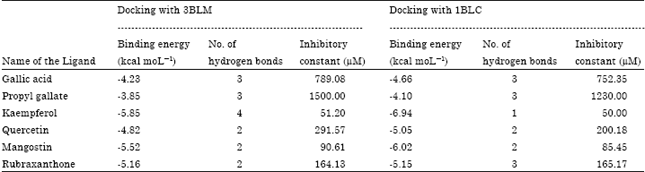 | |
These complexes were analyzed using the same package and the interactions involved in the active site of the docked structures were examined. Among the various binding mode in the active site, the best pose was selected by figuring out the minimum binding energy. The same predictions were done for all ligands and the most stable conformations were selected. Inhibitory constants of each ligand for 3BLM and 1BLC were also predicted. Among the ligands evaluated, kaempferol showed the binding energies of-5.85 and-6.94 kcal mol-1 with the significant inhibitory constants of 51.2 μM and 50 μM, when compare to the others for 3BLM and 1BLC, respectively (Table 3).
Comparison of dock made for 3BLM and 1BLC against Gallic acid revealed an interesting pattern of interaction. The binding energy of Gallic acid with 3BLM was found to be -4.23 kcal mol-1 and formed three hydrogen bonds with Gln237, Asn132 and Ser130, although the complex contained other interacting residues such as Gln236, Asn170, Ser235, Lys234 and Ser70 (Fig. 3 Ia). However, the binding energy of the same ligand with that of 1BLC active site was found to be -4.66 kcal mol-1 even though, it formed three hydrogen bonds with the same residues at the similar positions while the other residues (Tyr105, Ser70) interacting were revealed to be different with the former (Fig. 3 Ib). The binding energies of docking Propyl gallate was found to be -3.85 and -4.1 kcal mol-1 with 3BLM and 1BLC, respectively, however, both formed only two hydrogen bonds but at different positions viz., Asn170 and Gln237 for 3BLM and at Asn214 and Lys73 for 1BLC, respectively. Perhaps, the other residues interacting with Propyl gallate were also identified to be different for both (Ile167, Tyr105, Asn132, Glu166, Ala 69 for 3BLM and Ser235, Met127, Gln237, Gly236 for 1BLC) except Ser70 (Fig. 3 IIa, b).
Kaempferol successfully docked with 3BLM by forming four hydrogen bonds at Asn170, Glu166, Arg244 and Gln237 and exhibited the binding energy of -5.85 kcal mol-1 although it interacted with other residues at positions such as Asn132, Ser70, Ser130 and Ser235 (Fig. 3 IIIa). The same with that of 1BLC was found to be -6.94 kcal mol-1 and unlike that of 3BLM formed just a single hydrogen bond with Lys73; however the structure also exhibited interaction with Asn132, Ser70 and Ser130 (Fig. 3 IIIb). The binding energy of Quercetin that docked with active site of 3BLM and 1BLC was found to be -4.82 and -5.05 kcal mol-1, respectively. It formed two hydrogen bonds with Arg244 and Ser235 by 3BLM and with Lys73 and Ser126 by 1BLC (Fig. 3 IVa, b). Other residues such as Ser130, Ile167, Gln237 and Asn170 interacting with Quercetin was found to be in common for both, however it differed with respect to residues Tyr105, Ser70, Asn132 for 3BLM and Glu166, Gly236 and Ser235 for 1BLC, respectively.
The binding energy of docking Mangostin with 3BLM and 1BLC active sites was found to be -5.52 and 6.02 kcal mol-1, respectively. It formed two hydrogen bonds with Ser235 and Glu166 with 3BLM and each one with Ser235 and Lys73 for 1BLC, respectively.
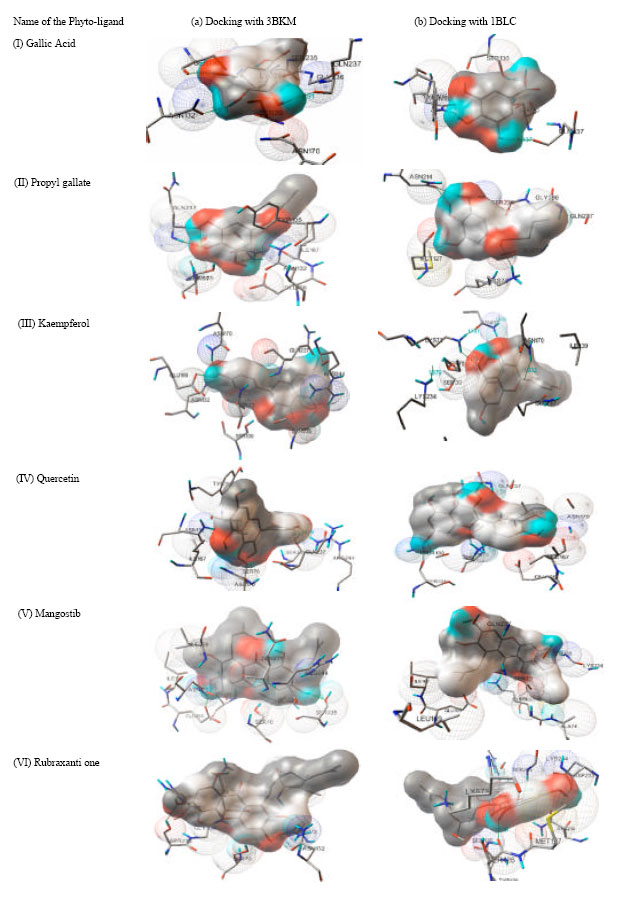 | |
| Fig. 3 (a-b): | Docking interactions of the phyto-ligands with beta-lactamases of S. aureus |
Other residues interacting were Ser70, Ile239, Asn132, Asn170, Ile167 and Arg244 for 3BLM and Gln237, Leu169, Glu66, Ile167, Ser70 and Lys234 for 1BLC, respectively (Fig. 3 Va, b). The binding energies of docking Rubraxanthone with 3BLM and 1BLC active site was found to be -5.16 and -5015 kcal mol-1 respectively which formed two hydrogen bonds each with Asn170 and Ser235 for 3BLM and three hydrogen bonds, one each with Asp233, Ser235 and Ser126 for 1BLC, respectively. Other interacting residues were found to be Ser70, Ile239, Asn132 and Arg244 with 3BLM and Arg244, Lys73, Ser130, Tyr129, Asn124 and Lys234 with 1BLC (Fig. 3 VIa, b).
Among the six ligands screened, Kaempferol was found to have the best docking in terms of both binding affinity and strong hydrogen bond interactions. The minimum binding energy indicated the strong affinity of the molecule with the receptor which had been stabilized by strong hydrogen bond interactions in the binding pocket although, the efficiency of other ligands was also checked and the corresponding inhibitory constants were predicted. This information suggests that all the six natural compounds used in the present study could be considered as lead compounds for further analysis.
DISCUSSION
The frenzied researches on the antibiotics during the last five decades have resulted in the production of more than ten thousand potential molecules from the microbes alone (Raja and Prabakaran, 2011). Given the structure of a biological receptor, it should be possible to design or discover molecules that will bind to it. Using atomic resolution structures and computational techniques, investigators have attempted to design or discover novel inhibitors for biological receptors. Such putative ligands have been selected for their complementarity to the structure of the receptor. Plants have an almost limitless ability to synthesize aromatic substances, most of which are phenols or secondary metabolites or their oxygen-substituted derivatives.
Development of inhibitors for β-lactamases could reduce the rate of resistance as they were designed to inhibit the catalytic activity of the enzyme. Although all these inhibitors are clinically tested and widely used, the new generation of AmpC β-lactamases and methicillin resistant strains in Staphylococcus aureus show resistance to these compounds. Since, natural products have a very good resource of biological systems which represent novel structures that can be considered as the drug candidates in the protein structural study (Costello et al., 2006), developing in silico models for understanding the relationship between important ADME parameters and molecular structure with properties could play a major role in identifying new drugs. However, in the present study, all the ligands were found to obey the above stated Lipinski Rule of five. Among the six ligands chosen Gallic acid, Propyl Gallate and Kaempferol perfectly obeyed all the five rules. Quercetin was found to have 6 donor atoms, whereas all other conditions were satisfied. Moreover, Rubraxanthone contained 6 rotatable bonds with 5.51 logP (solubility) value. Mangostin satisfied all the rules except for logP value as it was measured to be 5.35. Although these values lied in the range of values suggested by Ghose et al. (1999), the range of the modified values suggested that the molecular weight and logP should not exceed 480 Da and 5.6, respectively. Since, the current drug discovery tends toward the development of larger and more lipophilic molecules and a shift to higher molecular weight and lipophilicity is also apparent for the development of leads to drugs, all the ligands selected for the present study contained aromatic rings which ranged from 1-3 and that the structures of Rubraxanthone and Mangostin were very close to each other in almost all the properties and all the molecules considered with all the orbitals filled and with nullified charges.
A mechanism by which a protein receptor recognizes its ligand and its further interaction with those molecular substrates are of much importance. Association between a protein and a substrate allows us to predict and rank the structures on the basis of its binding and interaction (Sousa et al., 2006). Optimization of the coordinate geometries of the ligands followed by the docking process improved the docking accuracy and the binding energy (Hu and Shelver, 2003). A better knowledge of the structure of ligands improved the quality of docking by aligning themselves to the minimum energy-binding mode. Since previous researches show the similar kind of calculations give reliable conformations (Frison and Ohanessian, 2008), structures of the ligand were brought to its minimum energy state using GAMESS US quantum mechanical package.
Hartree-Fock methods give the easiest way to calculate the properties of an electronic structure. Polarisable Continuum Model (PCM) developed by Tomasi et al. (2005) was used to study the solvation effects on molecules that bear high formal charge. Here, the energy of the solvated state is not considered as they differ in polarity and size. Perhaps the binding affinity of a ligand for a receptor depends on the relative free energies in solution. Thus, novel ligands possess high charge without solvation which can give ambiguous binding positions at the ligand site of the protein (Shoichet et al., 1999). Hence, in this perception, in the present study, care was taken to bring in solvation effect by using PCM to the molecules in the dielectric constant of water. All the ligand molecules which were optimized in atmospheric environment, were subjected to have solvation effect.
The optimized energies of the ligands in both gas and water phase revealed to gain minimum values, compared to the non-optimized structures. For an instance, the energy of Gallic acid in non-optimized state was calculated as -638.4187722550 A.U and the same after solvation effect got more polarized charges and gave value of -638.4956284154 which in turn was less than the calculated energy (0.0768561604 A.U) of gas phase. Hence, the change in the energy values indicated the changes in the structural parameters of the ligands. The bond lengths of flexible bonds especially hydrogen bond donors and acceptors in the ligand structures were calculated unlike the rigid bonds such as, alkyl group as they do not have hydrophilic properties. In most of the ligand structures, the bond lengths of carbonyl groups were increased except for Propyl gallate, due to the fact that an increase in charge separation increased the polarization of carbonyl groups making the structure more stable for its utility as an agent for the target (β-lactamase) in docking analysis.
A drug molecule is triggered when the binding of small molecule to the receptor protein is perfectly done. Such protein-ligand interaction is comparable to the lock-and-key principle, in which the lock encodes the protein and the key is ensembled by the ligand. The major driving force for binding appears to be hydrophobic interaction whose specificity is however controlled by hydrogen bonding interactions (Kubinyi, 1998). Analysis of the six ligands to active site of 3BLM and 1BLC showed similar binding modes and interactions with the residues. The receptors were kept rigid throughout docking while the ligands were allowed to rotate freely in the active site to achieve suitable conformations. Autodock4 generated 100 docking simulations in the active site of the ligands and among those the most stable orientation was chosen according to the value of binding energy. The most stable conformation possessed minimum energy which explained the mechanism by which the ligand occupied the active site area as proved by Alberts et al. (2005).
Even though docking with 3BLM and 1BLC showed similar interactions with all the six ligands, the number of active site residues participating in strong interactions with the ligands was found to be different. Perhaps, the reason behind this could be explained on the basis of the conformational changes in 1BLC, when it bound with clavulanic acid. Moreover, residues in the active site of 3BLM were more flexible to the ligands, when compared to those in 1BLC. Hence, the docking of 3BLM with six ligands is considered in this context. Gallic acid which was the smallest ligand among the six, docked successfully against both structures however, the interacting residues were found to be the same. The reduced size of Gallic acid made it to fit in the binding site pocket easily. Among the three hydrogen bonds formed, SER 130 was found to be more interesting as it was one among the catalytic residues in 3BLM.
Docking of 3BLM and 1BLC with other five ligands did not show much similarity in the interacting residues, as the ligand size was increasing. In 3BLM-Propyl Gallate docking analysis the binding energy was found to be -3.85 kcal mol-1 and the number of hydrogen bond interactions were two. None of the catalytic residues took part in these interactions. Moreover, Kaempferol exhibited minimum binding energy value (-5.85 kcal mol-1) and formed four hydrogen bonds with 3BLM, in which GLU 166 was given more importance as it belonged to catalytic residues.
While considering other ligands such as Quercetin, Mangostin and Rubraxanthone, the second one showed minimum binding energy of -5.52 kcal mol-1 with 3BLM and formed two hydrogen bonds with SER 235 and GLU 166. Another efficient docking was observed with Rubraxanthone, whose binding energy was -5.16 kcal mol-1, though it formed no hydrogen bonds with catalytic residues. Quercetin showed a weak interaction with 3BLM when compared to Mangostin and Rubraxanthone and proved the binding energy of -4.82 kcal mol-1 and had no interaction with any of catalytic domain at the active site. If compounds binding to the target proteins are known, the three dimensional structures of their complexes with the target protein are of interest because, they may give hints about the catalytic mechanism of the enzyme or opportunities for optimizing the binding affinity of the compound (Mannhold et al., 2002). Accordingly, the results of the present investigation gave a clear indication that all six ligands could be considered as an efficient lead for binding 3BLM, however, Kaempferol could be considered as a good inhibitor of β-lactamase since, it gave minimum binding energy with maximum number of interactions compared to the others. Moreover, Kaempferol has been reported for the inhibitory activity against β-lactamase in Methicillin-Resistant Staphylococcus aureus (MRSA). Our study in-fact, gives support to the fact that this compound could be considered as an effective drug in this field with intensive work on its pharmacodynamic and pharmacokinetic understanding.
CONCLUSION
Antibiotic resistance has turn out to be a worldwide public health crisis that continues to grow up. Development of inhibitors for β-lactamases could reduce the rate of resistance as they are designed to inhibit the catalytic activity of the enzyme. Inhibitors generally have little antimicrobial properties themselves and so are combined with a beta-lactam antibiotic. Although all these inhibitors are clinically tested and widely used, the new generation of AmpC β-lactamases and methicillin resistant strains in Staphylococcus aureus shows resistance to these compounds. As, classical inhibitors designed fail to act efficiently in blocking the catalytic property of β-lactamases, necessitates the design of novel inhibitors. Thus, the present study was focused on evaluating the inhibitory activity of natural compounds such as Gallic acid, Propyl Gallate, Kaempferol, Quercetin, Mangostin and Rubraxanthone over in silico biology. Kaempferol was identified to be the best ligand as it formed four hydrogen bonds with minimum inhibitory constant values, when compared with the other ligands.
REFERENCES
- Alberts, I.L., N.P. Todorov and P.M. Dean, 2005. Receptor flexibility in de novo ligand design and docking. J. Med. Chem., 48: 6585-6596.
PubMedDirect Link - Aqil, F., M.S. Khan, M. Owais and I. Ahmad, 2005. Effect of certain bioactive plant extracts on clinical isolates of β-lactamase producing methicillin resistant Staphylococcus aureus. J. Basic Microbiol., 45: 106-114.
PubMed - Chen, C.C. and O. Herzberg, 1992. Inhibition of β-lactamase by clavulanate: Trapped intermediates in cryocrystallographic studies. J. Mol. Biol., 224: 1103-1113.
PubMed - Costello, A.L., N.P. Sharma, K.W. Yang, M.W. Crowder and D.L. Tierney, 2006. Ray absorption spectroscopy of the zinc-binding sites in the class B2 metallo-β-lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry, 45: 13650-13658.
PubMed - Crisostomo, M.I., H. Westh, A. Tomasz, M. Chung, D.C. Oliveira and H. de Lencastre, 2001. The evolution of methicillin resistance in Staphylococcus aureus: Similarity of genetic backgrounds in historically early methicillin susceptible and resistant isolates and contemporary epidemic clones. Proc. Natl. Acad. Sci. USA., 98: 9865-9870.
PubMed - Ekins, S., C.L. Waller, P.W. Swaan, G. Cruciani, S.A. Wrighton and J.H. Wikel, 2000. Progress in predicting human ADME parameters in silico. J. Pharm. Toxicol. Methods, 44: 251-272.
CrossRef - Fan, X., Y. Liu, D. Smith, L. Konermann, K.W. Siu and D. Golemi-Kotra, 2007. Diversity of penicillin-binding proteins: Resistance factor FmtA of Staphylococcus aureus. J. Biol. Chem., 282: 35143-35152.
PubMed - Frison, G. and G. Ohanessian, 2008. A comparative study of semiempirical, ab initio and DFT methods in evaluating metal-ligand bond strength, proton affinity and interactions between first and second shell ligands in Zn biomimetic complexes. J. Comput. Chem., 297: 416-433.
PubMed - Georgopapadakou, N.H., 1993. Penicillin-binding proteins and bacterial resistance to β-lactams. Antimicrob. Agents Chemother., 37: 2045-2053.
PubMed - Ghose, A.K., V.N. Viswanadhan and J.J. Wendoloski, 1999. A knowledge based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. J. Comb. Chem., 113: 55-68.
PubMed - Gohlke, H. and G. Klebe, 2001. Statistical potentials and scoring functions applied to protein-ligand binding. Curr. Opin. Struct. Biol., 1136: 231-235.
PubMed - Gold, H.S. and R.C. Moellering, 1996. Antimicrobial-drug resistance. N. Engl. J. Med., 335: 1445-1453.
Direct Link - Herzberg, O., 1991. Refined crystal structure of beta-lactamase from Staphylococcus aureus PC1 at 2.0 A resolution. J. Mol. Biol., 217: 701-719.
PubMed - Hu, X. and W.H. Shelver, 2003. Docking studies of matrix metalloproteinase inhibitors: Zinc parameter optimization to improve the binding free energy prediction. J. Mol. Graph. Model, 22: 115-126.
PubMed - Krejsa, C.M., D. Horvath, S.L. Rogalski, J.E. Penzotti, B. Mao, F. Barbosa and J.C. Migeon, 2003. Predicting ADME properties and side effects: The BioPrint approach. Curr. Opin. Drug Discov. Dev., 649: 470-480.
Direct Link - Kubinyi, H., 1998. Structure-based design of enzyme inhibitors and receptor ligands. Curr. Opin. Drug Discov. Dev., 1: 4-15.
PubMed - Lawson, S.G., T.L. Mason, R.D. Sabin, M.E. Sloan, R.R. Drake, B.E. Haley and B.P. Wasserman, 1989. UDP-Glucose: (1,3)-β-glucan synthase from Daucus carota L.: Characterization, photoaffinity labeling and solubilization. Plant Physiol., 90: 101-108.
PubMed - Li, X.Z., D. Ma, D.M. Livermore and H. Nikaido, 1994. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: Active efflux as a contributing factor to β-lactam resistance. Antimicrob. Agents Chemother., 38: 1742-1752.
PubMed - McLaughlin, J.R., C.L. Murray and J.C. Rabinowitz, 1981. Unique features in the ribosome binding site sequence of the gram-positive Staphylococcus aureus β-lactamase gene. J. Biol. Chem., 256: 11283-11291.
PubMed - Padayatti, P.S., M.S. Helfand, M.A. Totir, M.P. Carey, P.R. Carey, R.A. Bonomo and F. van den Akker, 2005. High resolution crystal structures of the trans-enamine intermediates formed by sulbactam and clavulanic acid and E166A SHV-1 β-lactamase. J. Biol. Chem., 280: 34900-34907.
PubMedDirect Link - Raja, A. and P. Prabakarana, 2011. Actinomycetes and drug-An overview. Am. J. Drug Discovery Dev., 1: 75-84.
CrossRefDirect Link - Rotschafer, J.C. and B.E. Ostergaard, 1995. Combination β-lactam and β-lactamase inhibitor products: Antimicrobial activity and efficiency of enzyme inhibition. Am. J. Health Syst. Pharm., 52: S15-S22.
PubMed - Sandanayaka, V.P. and A.S. Prashad, 2002. Resistance to β-lactam antibiotics: Structure and mechanism based design of β-lactamase inhibitors. Curr. Med. Chem., 9: 1145-1165.
PubMed - Shoichet, B.K., A.R. Leach and I.D. Kuntz, 1999. Ligand solvation in molecular docking. Proteins, 34: 4-16.
PubMed - Sousa, S.F., P.A. Fernandes and M.J. Ramos, 2006. Protein-ligand docking: Current status and future challenges. Proteins, 65: 15-26.
PubMed - Tomasi, J., B. Mennucci and R. Cammi, 2005. Quantum mechanical continuum solvation models. Chem. Rev., 105: 2999-3093.
PubMed