
Mustafa Abdo Saif Dehwah
College of Life Sciences, Hua Zhong Normal University, Wuhan, Hubei 430079, Peoples Republic of China
Zhang Shuang
College of Life Sciences, Hua Zhong Normal University, Wuhan, Hubei 430079, Peoples Republic of China
Wang Zhen Hua
College of Life Sciences, Hua Zhong Normal University, Wuhan, Hubei 430079, Peoples Republic of China
Wang Min
College of Life Sciences, Hua Zhong Normal University, Wuhan, Hubei 430079, Peoples Republic of China
Qing-Yang Huang
College of Life Sciences, Hua Zhong Normal University, Wuhan, Hubei 430079, Peoples Republic of China
Journal of Applied Sciences
Year: 2009 | Volume: 9 | Issue: 19 | Page No.: 3407-3423







ABSTRACT
The aim of this review was to discuss the current understanding of Type 2 Diabetes Mellitus (T2DM) genetic advance and aetiology. The T2DM is a genetically heterogeneous disease, with several relatively rare monogenic forms and a number of more common forms resulting from a complex interaction of genetic and environmental factors. Earlier studies using a candidate gene approach, family linkage studies and gene expression profiling uncovered a number of T2DM genes, but the genetic basis of common T2DM remained unknown. Recent years have seen a tremendous surge in our understanding of the genetics of T2DM by Genome-Wide Association Study (GWAS). Approximately 20 genes consistently associated with T2DM mainly implicate pancreatic β-cell function in the pathogenesis of T2DM.
PDF Abstract XML References Citation
How to cite this article
Mustafa Abdo Saif Dehwah, Zhang Shuang, Wang Zhen Hua, Wang Min and Qing-Yang Huang, 2009. Type 2 Diabetes: Genetic Advance and Aetiology. Journal of Applied Sciences, 9: 3407-3423.
DOI: 10.3923/jas.2009.3407.3423
URL: https://scialert.net/abstract/?doi=jas.2009.3407.3423
DOI: 10.3923/jas.2009.3407.3423
URL: https://scialert.net/abstract/?doi=jas.2009.3407.3423
INTRODUCTION
Type 2 Diabetes Mellitus (T2DM) is a complex heterogeneous group of metabolic condition characterized by elevated levels of serum glucose, caused mainly by impairment in both insulin action and insulin secretion. T2DM is a complex trait where common genetic variants having modest individual effects act together and interact with environmental factors to modulate the risk of the disease. Traditional genetic research, including twin, adoption, and family studies, consistently supports that T2DM has a genetic component. Two broad approaches have been used to define the genetic predisposition of T2DM. First, molecular events in T2DM pathogenesis have been examined directly by testing the role of sequence variants of specific candidate genes. The candidate gene approach focuses on the search for an association between T2DM and sequence variants in or near biologically defined candidate genes which have been chosen based on their known physiological function. To overcome the shortcomings of the candidate gene studies, investigators have applied a genome-wide linkage scan strategy in which regularly spaced markers are traced in families and sibling pairs for segregation with T2DM. No prior knowledge of gene or gene effects is necessary, but the genetic locus must have sufficient impact on the disease susceptibility to be detectable. During the past decades, extensive efforts have been made to detect the underlying genetic structure for T2DM. However, until very recently, the genes involved have been poorly understood. Using a new and powerful technology in the form of a genome-wide chip that genotypes up to hundreds of thousands of SNPs, Genome-Wide Association Studies (GWAS) have recently led to the discovery of a group of novel genes that were reproducibly associated with T2DM risk. These studies have increased our understanding of the genetic aetiology of T2DM and provided invaluable insights into the way genetic studies should be conducted. The present review discusses the current understanding of T2DM genetic advance and aetiology.
EVIDENCE FOR THE GENETIC COMPONENT OF T2DM
The genetic component of T2DM comes from ethnic group differences in prevalence rates. These differences in prevalence range from 1% in Mapuche Indian tribes or Chinese population living in rural areas in mainland China, to extremely high levels found in Nauru and Pima Indians in Arizona (King and Rewers, 1993). Furthermore, the prevalence is higher in full-blooded Nauruan and Pima Indians than in those with admixture (Knowler et al., 1988; Serjeantson et al., 1983). Another source of evidence for genetic contribution in T2DM is familial aggregation. Lifetime risk of T2DM development is 40% in offspring of one T2DM parent and increases up to 70% if both parents have T2DM (Groop and Tuomi, 1997). In addition, evidence shows a greater likelihood of T2DM in offspring of affected mothers than affected fathers, indicating an excess maternal transmission of the disease (Thomas et al., 1994; De Silva et al., 2002; Arfa et al., 2007). Family, history of T2DM has a significant, independent and graded association with the prevalence of T2DM. In twins studies, very high concordance (55 to 100%) for T2DM has been reported among monozygotic (MZ) twins (Newman et al., 1987; Barnett et al., 1981).
Mutations in some genes cause rare forms of T2DM, giving additional support for the genetic roles in the aetiology of the disease. For example, genes KCNJ11 and ABCC8 carry rare mutations that cause a Mendelian form of neonatal diabetes (Babenko et al., 2006; Gloyn et al., 2004). Specific mutation in the mitochondrial genome was found to cause maternally inherited diabetes and deafness (Van Ouweland et al., 1994). Maturity-onset Diabetes of the Young (MODY), which is characterized by high penetrance, early age at onset of hyperglycemia and defective function of β-cells in the pancreas, accounts for 1-5% of all T2DM cases. Mutations in HNF4A, GCK, HNF1A, IPF1, HNF1B (TCF2) and NEUROD1 cause different subtypes of MODY1-6 (Fajans et al., 2001).
SEARCH FOR T2DM SUSCEPTIBILITY GENES
Susceptibility genes identified by linkage studies: The first T2DM gene identified by linkage is CAPN10 that belonged to the family of calcium activated non-lysosomal thiol proteinases. Horikawa et al. (2000) used relatively sparse Linkage Disequilibrium (LD) map of SNPs in the region of linkage on chromosome 2q to identify 3 common intronic variants of a previously unknown gene. Both a single intronic variant (UCSNP-43: G to A) and a specific haplotype combination defined by three polymorphisms (UCSNP-43, -19 and -63) were associated with T2DM in Mexican Americans, with lesser evidence of an association in a Northern European population from the Botnia region of Finland. Surprisingly, individuals with a combination of two different haplotypes were at the highest risk of T2DM (Horikawa et al., 2000; Wang et al., 2002). Baier et al. (2000) showed altered gene transcription and reduced muscle mRNA levels in muscle biopsies from Pima Indians with T2DM. CAPN10 SNP-44 (rs2975760 g4841T/C), located in intron 3 and 11 bp from SNP-43 (rs3792267 A/G), was independently associated with T2DM in several populations and was in LD with the missense mutation Thr504Ala and two 5'-UTR variants (SNP-134 and SNP-135). An initial meta-analysis also supported an association with T2DM (Weedon et al., 2003). Furthermore, CAPN10 variants were valuable along with traditional risk factors in predicting the onset of T2DM in a prospective study of Botnian Finnish individuals (Lyssenko et al., 2005).
The TCF7L2 gene encodes for an enteroendocrine transcription factor that has a role in the Wnt signaling pathway, which is one of the key developmental and growth regulatory mechanisms of the cell. Reynisdottir et al. (2003) found suggestive linkage of T2DM to chromosome 10q in the Icelandic population. Fine-mapping with 228 microsatellite markers in Icelandic individuals with T2DM and controls throughout a 10.5 Mb interval on 10q identified one microsatellite marker, DG10S478 in intron 3 of the TCF7L2 gene, as being strongly associated with T2DM (p = 2.1x10-9) (Grant et al., 2006). This association was replicated in both a Danish cohort (p = 4.8x10-3) and a US cohort (p = 3.3x10-9). Replications have appeared from analysis in subjects of Amish (Damcott et al., 2006), Finnish (Scott et al., 2006), UK samples (Groves et al., 2006), French (Cauchi et al., 2006), US (Zhang et al., 2006; Saxena et al., 2006), German KORA 500 K study population (Herder et al., 2008), population-based study of Caucasian (Van Hoek et al., 2008) and Chinese population (Ng et al., 2007; Chang et al., 2007). A meta-analysis of 28 original published association studies included 29,195 control subjects and 17,202 cases confirmed the association between the TCF7L2 rs7903146 polymorphism and susceptibility to T2DM (OR = 1.46 95% CI 1.42-1.51; p = 5.4x10-140). Compared with any other gene variants previously confirmed by meta-analysis, TCF7L2 can be distinguished by its tremendous reproducibility of association with T2DM and its OR twice as high (Cauchi et al., 2007). Very recently, a large meta-analysis summarizes the strong evidence for an association between TCF7L2 gene and T2DM both overall and in Caucasians, North Europeans, East Asians, Indians, and Africans and suggested a potential multiplicative genetic model for all the four polymorphisms (rs7903146, rs7901695, rs12255372, rs11196205) of TCF7L2 gene among different ethnic populations except for Africans, where additive genetic mode is suggested for rs7903146 polymorphism, as well as suggests the TCF7L2 gene involved in near 1/5 of all T2DM (Tong et al., 2009). The increase in risk of T2DM associated with the TCF7L2 variant alleles so far identified is substantially greater than that associated with variants in the other confirmed T2DM-susceptibility genes (PPARγ, KCNJ11). The strongest signal for T2DM in GWAS was found for TCF7L2 rs7903146 with p-values down to <10-48 and increase in the odds for the disease of 37% (Scott et al., 2007; Saxena et al., 2007). In the prediction study, TCF7L2 rs7903146 was significantly associated with the risk of future T2DM (OR = 1.30; p = 9.5x10-13) and predicted progression from normal glucose tolerance to T2DM (OR = 1.27; p = 2.7x10-7) in the MPP cohort (Lyssenko et al., 2008).
The locus at 18p was originally found to be linked to T2DM in families from Finland and Southern Sweden (Parker et al., 2001) and was confirmed in a second series derived from the Southwest of the Netherlands (Van Tilburg et al., 2003). The strongest evidence was obtained for marker D18S63 (LOD score 2.3, nominal p = 0.0006). In all three studies, the effect of the 18p region was strongest in the obese subpopulation. One of the human homologs of mouse Lpin1 gene, LPIN2, is located in the 18p11 region. LPIN2 is ubiquitously expressed in different tissues, including skeletal muscle (Zhou and Young, 2005). The LPIN2 gene is responsible for the lipodystrophy in the fld line, which, among other traits, is characterized by severe insulin resistance. Fdl mice, in which Lpin1 is deleted, have diminished adipose tissue mass and multiple pathologies, including insulin resistance, fatty liver and progressive neuropathy of peripheral nerves (Shimomura et al., 1999; Péterfy et al., 2001). The protein Lipin is required upstream of PPARγ for normal adipocyte differentiation (Phan et al., 2004). The rs3745012 SNP of the LPIN2 gene is associated with T2DM and fat distribution (Aulchenko et al., 2007).
The human PTPN1 gene maps on chromosome 20q13.13 which showed evidence of linkage with early onset T2DM in a subset of 55 French families (Zouali et al., 1997). Multiple noncoding SNPs in the PTPN1 gene were implicated in T2DM in Caucasian and Mexican-American populations (Bento et al., 2004; Palmer et al., 2004), with the most consistent evidence for association occurring with SNPs spanning the 3' end of intron 1 of PTPN1 through intron 8 (p = 0.043-0.002). All of the associated SNPs were present in a single 100 kb haplotype block that encompassed the PTPN1 gene. Haplotype frequencies were s ignificantly different between T2DM case and control subjects (p = 0.0035-0.0056), with a single common haplotype (36%) contributing strongly to the evidence for association, with OR=1.3. Furthermore, the same haplotypes were associated with glucose homeostasis measures in Hispanic subjects (Bento et al., 2004). However, Florez et al. (2005) failed to replicate the findings in a large Caucasian study.
Adiponectin (APMI) is a strong candidate for T2DM given the clear role of plasma adiponectin levels in insulin sensitivity and the fall in adiponectin levels with obesity and T2DM. Adiponectin was mapped to the region on chromosome 3q27 which found to have positive linkage to metabolic traits and T2DM (Vionnet et al., 2000; Hegele et al., 1999; Kissebah et al., 2000). Multiple studies found evidence that promoter variants in the APM1 gene were associated with T2DM in French and Swedish Caucasian and Japanese populations. However, APM1 variants did not appear to account for the 3q27 linkage in French families (Gibson and Froguel, 2004). APM1 variation at positions 45 (G allele) in exon 2 and 276 in intron 2 (T allele) were associated with a 4.5 fold increased risk of converting from impaired glucose tolerance to T2DM in the STOP-NIDDM trial (Zacharova et al., 2005). A meta-analysis of nine association studies included 2379 subjects confirmed significant association between the SNP45TG+GG and SNP276GG polymorphisms of APM1 with T2DM in Chinese populations (p = 0.05) (Li et al., 2008). The search for T2DM susceptibility genes on most other chromosomes (1q21.3-23, 2q37.3, 3p24.1, 3q28, 10q26.13, 12q24.31, and 18p11.22) is ongoing.
Susceptibility genes discovered by candidate gene association: Variants in genes encoding proteins that play a role in pathways involved in insulin control and glucose homeostasis are excellent candidates for T2DM. The candidate gene approach focuses on the search for an association between T2DM and sequence variants in or near biologically defined candidate genes which have been chosen based on their known physiological function. The importance of these variants is tested by comparing the frequency in T2DM subjects and control individuals. Numerous variants within several genes that confer an increased susceptibility to T2DM have been identified by candidate genes studies but only a small number have been identified as strong candidates for T2DM such as PPARγ and KCNJ11 genes, which they were confirmed by GWAS (Zeggini et al., 2007; Sladek et al., 2007).
The PPARγ is a member of nuclear hormone receptor superfamily of transcription factors and the target of the widely used class of insulin sensitizers, the thiazolidinediones. Multiple studies have examined a Pro12Ala polymorphism in the PPARγ2 isoform. The rare Ala allele is seen in about 15% of Europeans and was shown to be associated with increased transcriptional activity, increased insulin sensitivity and protection against T2DM in an initial study (Deeb et al., 1998). Subsequently, smaller studies provided inconsistent results. Larger studies have shown a consistently protective effect of the Ala12 allele when compared with the common Pro12 allele. The common Pro12 allele increases T2DM risk by an OR of 12.5 in a meta-analysis in cross sectional studies (Altshuler et al., 2000). Several, but not all studies suggest that the rare Ala12 allele protects against insulin resistance and obesity, but the association with obesity has been inconsistent. A meta-analysis of 41 published and 2 unpublished studies included 42,910 subjects (Asia, Europe and North America) demonstrated that the reduced risk of T2DM in Ala12 carriers is not homogeneous (Ludovico et al., 2007). Recent replication studies reported significant associations between rs17036328, rs11709077, rs1801282 in PPARγ2 and T2DM in the German KORA 500 K study population (Herder et al., 2008). PPARγ2 Pro12 allele was associated with T2DM in Hubei Han Chinese population (Dehwah et al., 2008). In the prediction study, PPARγ2 SNP rs1801282 was significantly associated with the risk of future T2DM (OR = 1.20; p = 4.0x10-4) and predicted progression from normal glucose tolerance to T2DM (OR = 1.15; p = 0.03) in the MPP cohort (Lyssenko et al., 2008).
KCNJ11 encodes a subunit of an inwardly rectifying ATP-sensitive potassium channel I (KATP). I (KATP) channels are crucial for the regulation of glucose-induced insulin secretion in pancreatic β-cell. The β-cell potassium channel comprises two subunits, the potassium channel encoded by the gene KCNJ11 and the regulatory subunit (SUR1 encoded by ABCC8) that binds sulfonylureas and ATP. Variants in both subunits have been associated with T2DM. Two variants in the ABCC8 gene were initially associated in smaller studies (Inoue et al., 1996; Hart et al., 1999; Meirhaeghe et al., 2001) and larger subsequent studies appeared to confirm the association (Barroso et al., 2003; Hani et al., 1997). However, the other studies have not confirmed the association (Gloyn et al., 2003; Florez et al., 2004). The association between these variants and altered insulin secretion was also observed (Elbein et al., 2001; Hansen et al., 1998). The adjacent KCNJ11 gene was initially noted to have multiple nonsynonymous coding variants (Inoue et al., 1997), but none were associated with T2DM (Altshuler et al., 2000; Sakura et al., 1996). However, a E23K polymorphism was shown to lower the sensitivity of the potassium channel to ATP and thus reduce insulin secretion in vitro (Schwanstecher et al., 2002) and to be associated with T2DM (Barroso et al., 2003; Love-Gregory et al., 2003; Yokoi et al., 2006). The result of Meta-analysis confirmed the association between the E23K variant and susceptibility to T2DM in UK cohort (K allele OR = 1.23; p = 1.5x10-5; KK genotype OR = 1.65; p = 2x10-6) (Gloyn et al., 2003) and white case-control subjects (n = 2,824, OR = 1.49, p = 2.2x10-4) (Nielsen et al., 2003). Subsequently, Florez et al. (2004) thoroughly examined KCNJ11 and ABCC8 and tested sufficient variants to completely tag all variation in this region. Again, the E23K variant was associated with T2DM in 3,413 subjects and the association was confirmed in a meta-analysis that included over 5000 T2DM subjects and 4747 controls (p<10-5, OR = 1.15). Recent replication studies reported significant associations between common variants in the KCNJ11genes and T2DM in a Japanese population (Omori et al., 2008).
Evidence for a role of the HNF4A gene in T2DM predisposition is also mounting (Barroso et al., 2003; Love-Gregory et al., 2004; Silander et al., 2004; Zhu et al., 2003). HNF4A regulates genes involved in glucose and fatty acid metabolism, as well as insulin secretion, and is therefore critical for maintaining lipid and glucose homeostasis. The K121Q variant of the ENPP1 gene has been best studied and shown to be associated with obesity but not T2DM in Caucasian and African American subjects ascertained from the New York Cancer Project (Matsuoka et al., 2006). Other investigators found an association of K121Q with earlier onset T2DM and coronary disease among T2DM patients (Bacci et al., 2005). The association of K121Q with T2DM was replicated in three relatively small populations, two of South Asian ancestry and one Caucasian (Abate et al., 2001).
Minton et al. (2002) sequenced DNA from 29 patients and uncovered 12 SNPs of WFS1. The most abundant genetic variant alters the amino acid at position 611 from a histidine to an arginine (H611R). The arginine variant was present in 40% of diabetic cases and 45% of controls (p<0.02), suggesting that the variant was protective from T2DM. Through large-scale candidate-pathway study, 1536 SNPs in 84 candidate genes were studied for association with T2DM, only (rs10010131, rs6446482, rs752854, rs734312) SNPs in WFS1 were associated with T2DM in UK and an Ashkenazi Jewish population (OR = 0.92 95% CI 0.88-0.95, p<10-4-10-7) (Sandhu et al., 2007). The result was then replicated in northern Swedish populations, the minor allele (G) at SNP rs752854 was statistically associated with reduced risk of T2DM (OR = 0.85 95% CI 0.75-0.96, p = 0.01), while borderline statistical significance was observed for the other three SNPs (rs10010131, rs6446482, rs734312) (Franks et al., 2008). Recent replication studies reported significant associations between WFS1 rs10012946 (proxy for rs10010131, r2 = 1.0) and T2DM in population-based study of Caucasian (Van Hoek et al., 2008). A meta-analysis of 11 association studies included 12,979 cases subjects and 14,937 controls was robustly confirmed the association of WFS1 rs10010131 with risk of T2DM (OR = 0.89 95% CI 0.86–0.92; p = 4.9x10-11) (Franks et al., 2008). In the prediction study, WFS1 SNP rs10010131 was significantly associated with the risk of future T2DM (OR = 1.12; p = 0.001) and predicted progression from normal glucose tolerance to T2DM (OR = 1.13; p = 0.004) in the MPP cohort (Lyssenko et al., 2008).
The HNF1B (also known as TCF2) gene is located on chromosome 17cen-q21.3, a region linked to T2DM (Demenais et al., 2003). Although, HNF1B is important for the development of the pancreas, mutations in HNF1B show other more characteristic phenotypes like cystic kidney disease, liver dysfunction and abnormal urogenital tract development (Nishigori et al., 1998; Lindner et al., 1999; Bingham et al., 2000). Polymorphisms in HNF1B were reported to be associated with T2DM in Caucasians (Bonnycastle et al., 2006; Winckler et al., 2007; Gudmundsson et al., 2007). However, the previously T2DM associated SNPs in HNF1B is not associated with T2DM in cohorts of 2,293 individuals from the Botnia study (Finland) and in 15,538 individuals from the Malmö Preventive Project (Sweden) (Holmkvist et al., 2008).
Novel susceptibility genes from genome wide association studies: Using a candidate gene approach, family linkage studies and gene expression profiling uncovered a number of T2DM susceptibility genes (Huang et al., 2006). Since, 2007, a new window has opened on defining potential T2DM genes through genome-wide SNP association (GWA) studies of very large populations of individuals with T2DM. GWAS revealed new susceptibility loci for T2DM and validated some of the known candidates (Table 1, Fig. 1). To date, the total of the twelve update GWA scans for T2DM have been published (Table 2). Six of these represent high-density scans (i.e., at least 300 000 SNPs, offering genome- wide coverage >65%), in samples of Northern European descent (Sladek et al., 2007; Saxena et al., 2007; Scott et al., 2007; Steinthorsdottir et al., 2007; Zeggini et al., 2007; Salonen et al., 2007). Another four studies featured a wider array of ethnic groups including Native American (Hanson et al., 2007), Hispanic (Hayes et al., 2007) and populations of European descent (Rampersaud et al., 2007; Florez et al., 2007), but were less extensive with respect to both sample size and SNP density (all used the Affymetrix 100K array). Two recent studies in East Asian subjects were on a smaller scale (featuring between 82 000 and 207 000 typed SNPs in a few hundred cases only) (Yasuda et al., 2008; Unoki et al., 2008).
The first GWAS covered 392,935 SNPs (passing quality control) and identified four novel loci including SLC30A8, LOC387761, IDE-KIF11-HHEX and EXT2-ALX4 (Sladek et al., 2007). Subsequence three GWAS analyzed 386,731, 393,453, and 315,635 SNPs respectively (Zeggini et al., 2007; Saxena et al., 2007; Scott et al., 2007). Common variants in CDKAL1, IGF2BP2, CDKN2A/B genes were significantly associated with T2DM risk, with the allele-associated odds ratios ranging from 1.07 to 1.48. These studies also confirmed the T2DM effects of SLC30A8 and HHEX. In another GWAS, Steinthorsdottir et al. (2007) found that variant rs7756992 in the CDKAL1 gene was significantly associated with T2DM risk in individuals of European ancestry (allele-specific OR = 1.20 95% CI 1.13-1.27) and Han Chinese ancestry (OR = 1.25 95% CI 1.11-1.40), but not in those of African ancestry.
| Table 1: | T2DM susceptibility loci for which there is genome-wide significant evidence for association* |
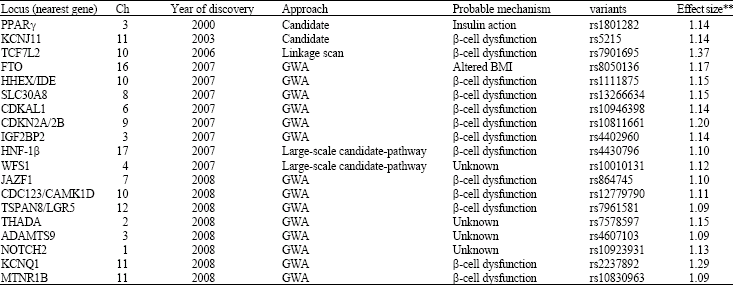 | |
| *ADAMTS9: ADAM metallopeptidase with thrombospondin type 1 motif 9; CAMK1D: Calcium/calmodulin-dependent protein kinase 1D; CDC123: Cell division cycle 123 homologue (Saccharomyces cerevisiae); CDKAL1: CDK5 regulatory subunit-associated protein1-like1; CDKN2A/2B: Cyclin-dependent kinase inhibitor 2A/2B; FTO: Fat mass and obesity associated; HHEX: Haematopoietically expressed homeobox; HNF1B: Hepatocyte nuclear factor 1 homeobox B; IDE: Insulin degrading enzyme; IGF2BP2: Insulinlike growth factor 2 mRNA binding protein 2; JAZF1: Juxtaposed with another zinc finger gene 1; KCNJ11: Potassium inwardly rectifying channel, subfamily J, member 11; KCNQ1: Potassium voltage-gated channel, KQT-like subfamily, member 1; LGR5: Leucine-rich repeat-containing G-protein coupled; NOTCH2: Notch homologue (Drosophila); PPARγ: Peroxisome proliferator-activated receptor gamma; SLC30A8: Solute carrier family 30 (zinc transporter), member 8; TCF7L2: Transcription factor 7 like 2; THADA: Thyroid adenoma associated; TSPAN8: Tetraspanin 8; WFS1: Wolfram syndrome1; MTNR1B: **Melatonin receptor 1B. Estimates of effect size (given as per-allele odds ratios) reported for European descent populations based on available data (Fig. 1) | |
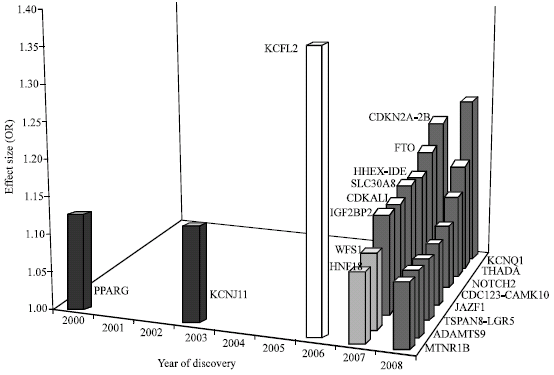 | |
| Fig. 1: | Effect sizes of 19 common T2DM susceptibility loci. The x axis gives the year that published evidence reached the levels of statistical confidence that are now accepted as necessary for genetic association studies. The y-axis gives the per-allele odds ratio (estimated for European-descent samples) for each locus listed on the y-axis. Loci are sorted by descending order of per-allele effect size from TCF7L2 (1.37) to ADAMTS9 (1.09) (Table 1). Loci shown in white and dark grey are those identified by GWA approaches and by Linkage scan respectively, whereas those identified by candidate-gene approaches and by large-scale candidate-pathway studies are shown in black and light grey, respectively. Odds ratios are as in Refs (Zeggini et al., 2007; Scott et al., 2007; Frayling et al., 2007; Franks et al., 2008; Zeggini et al., 2008; Yasuda et al., 2008; Prokopenko et al., 2009) |
| Table 2: | GWA scans for type 2 diabetes* |
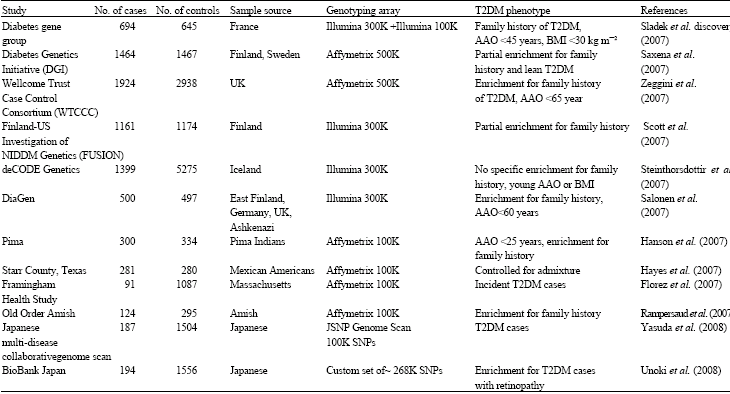 | |
| *AAO: Age at onset | |
Salonen et al. (2007) analyzed 315,917 HapMap-derived tagging SNPs in a two-stage study of 3,073 T2DM cases and 3,273 healthy controls. SNPs in the AHI1-LOC441171 region were found to confer ~30% increased risk. Other three GWAS genotyping 100 k SNPs in the Framingham Heart Study (Florez et al., 2007), Amish (Rampersaud et al., 2007) and American Indians (Hanson et al., 2007) reported some genetic variants related to the risk of T2DM. However, these associations were not univocally observed. Yasuda et al. (2008) reported that the C allele of SNP rs2237892 in the KCNQ1 gene is associated with an increased risk for developing T2DM in Japanese population. It is surprising that rs2237892 was associated with T2DM not only in the original Japanese set of cases and controls, but also in Chinese, Korean and European samples. At the same time Unoki et al. (2008) found similar evidence for SNPs closely related to rs2237892 (rs2237895 and rs2237897) in the KCNQ1 gene were strongly associated with T2DM in the Singaporean population of East Asian descent and the Danish population of European descent. These studies have demonstrated how studies in non-European descent populations can reveal novel susceptibility loci.
Strong signals for T2DM were observed for FTO, which is also highly associated with fat mass and obesity (Dina et al., 2007). The effect of FTO variants on T2DM risk has been replicated and seems to be mediated entirely by their marked effect on adiposity (Frayling et al., 2007). Melatonin receptor 1B (MTNR1B) gene is located on human chromosome 11q21-q22 and encodes one of two high affinity forms of a receptor for melatonin, the primary hormone secreted by the pineal gland. This gene product is an integral membrane protein that is a G-protein coupled 7-transmembrane receptor, its predominant expression in retina and brain (Reppert et al., 1995). GWAS have shown that variation in MTNR1B is associated with insulin and glucose concentrations. One of the strongest signals for glucose-stimulated insulin s ecretion in the DGI scan emanated from a SNP (rs10830963) in MTNR1B (p = 7x10-4, rank order 595) (Saxena et al., 2007). Given that the melatonin pathway had previously been suggested to be involved in pathogenesis of T2DM, the MTNR1B gene was a prime candidate gene for T2DM. The strong signal was observed at rs10830963, where each G allele was associated with an increase of (OR = 0.07 95% CI 0.06-0.08) mmol L-1 in fasting glucose concentrations (p = 3.2x10-50) and reduced β-cell function as measured by homeostasis model assessment in ~24,000 participants from the ten studies (HOMA-B, p = 1.1x10-15). The same allele was associated with an increased risk of T2DM (OR = 1.09 95% CI 1.05-1.12; per G allele p = 3.3x10-7) in large-scale meta-analysis of 13 case-control studies including data from GWAS, totaling 18,236 cases and 64,453 controls (Prokopenko et al., 2009). The rs10830963 variants in MTNR1B seem to have a more marked effect on risk of T2DM, the effect size being comparable in magnitude (OR = 1.09 95% CI 1.05-1.12) to several other T2DM susceptibility genes recently identified in GWAS. Lyssenko et al. (2009) provided strong support for a role of melatonin and its receptor MTNR1B in the pathogenesis of T2DM. A common variant in the MTNR1B receptor was associated with an increase in fasting glucose over time and predicted future T2DM, most likely through impairment of insulin secretion from the pancreatic β-cell function. The result of GWA meta-analysis for fasting plasma glucose indicated that the MTNR1B rs1387153 strongly modulates fasting plasma glucose in the European population (β = 0.06 mmol L-1, p = 7.6x10-29, n = 16,094) and increases the risk for T2DM (OR = 1.15 95% CI 1.08-1.22; p = 6.3x10-5, cases n = 6,332) (Bouatia-Naji et al., 2009). Effects of melatonin are mediated by two distinct receptors, MTNR1A and MTNR1B (Pandi-Perumal et al., 2008), which are members of the G-protein coupled receptor family, specifically inhibitory G-proteins (Gi). Both receptors have been found to be expressed in human and rodent islets (Muhlbauer and Peschke, 2007), with MTNR1A predominating, especially in glucagon-producing α-cells (Ramracheya et al., 2008). There is some evidence that melatonin may exert an effect on insulin secretion, in that acute effects exerted by cAMP-elevating agents are inhibited by melatonin, whereas prolonged effects of the hormone may be stimulatory (Peschke, 2008).The recent replication studies confirmed significant associations between SNPs within the HHEX, CDKN2A/B, CDKAL1 and KCNQ1 genes and T2DM in the Korean population (Lee et al., 2008), HHEX/KIF11/IDE, CDKN2A/B and IFG2BP2 loci in Danish subjects (Grarup et al., 2007), CDKAL1, IGF2BP2, CDKN2A/B, HHEX and SLC30A8 genes and T2DM in a Japanese population (Omori et al., 2008), CDKAL1, IGF2BP2, HHEX and FTO genes and T2DM in German KORA 500 K study population (Herder et al., 2008), ADAMTS9, CDKAL1, CDKN2A/B, FTO, IGF2BP2, JAZF1 and SLC30A8 in population-based study of Caucasian (Van Hoek et al., 2008), CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8 and KCNQ1 genes independently or additively contribute to T2DM risk in the Chinese Han population (Wu et al., 2008; Hu et al., 2009; Liu et al., 2008, 2009), TCF7L2 (rs12255372), CDKAL1 (rs7756992, rs7754840), HHEX (rs7923837), IGF2BP2 (rs4402960 and rs1470579), CDKN2A/B (rs10811661), and SLC30A8 (rs13266634) were significantly associated with T2DM in Japanese population (Tabara et al., 2009). The association of SLC30A8, HHEX, CDKAL1, CDKN2A/CDKN2B, IGF2BP2 and FTO with risk for T2DM was confirmed, in the recent large-scale study of Asian ancestry from Hong Kong and Korea, with odds ratios ranging from 1.13 to 1.35 (1.3x10-12<punadjusted<0.016) (Ng et al., 2008). It’s very recently reported that the common variants in the CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2, LOC387761 and CDKN2B genes did not confer a significant risk for T2DM in Pima Indians (Rong et al., 2009). In the prediction study, common variants in TCF7L2 (rs7903146), PPARγ (rs1801282), FTO (rs9939609), KCNJ11 (rs5219), NOTCH2 (rs10923931), WFS1 (rs10010131), CDKAL1 (rs7754840), IGF2BP2 (rs4402960), SLC30A8 (rs13266634), JAZF1 (rs864745, HHEX (rs1111875) were significantly associated with risk of future T2DM (OR = 1.30; p = 9.5x10-13, OR = 1.20; p = 4.0x10-4, OR = 1.14; p = 9.2x10-5, OR = 1.13; p = 3.6x10-4, OR = 1.13; p = 0.02, OR = 1.12; p = 0.001, OR = 1.11; p = 0.004, OR = 1.10; p = 0.008, OR = 1.10; p = 0.008, OR = 1.08; p = 0.03 and OR = 1.07; p = 0.03, respectively) in the MPP cohort (Lyssenko et al., 2008).
Additional loci from Meta analysis: Efforts to find additional T2DM susceptibility loci have to contend with the modest effect sizes anticipated and the stringent significance thresholds required when many hundreds of thousands of SNPs are tested in parallel. The obvious solution is to increase sample size and the most effective strategy for this involves combining existing GWAS data through meta-analysis. The Diabetes Genetics Replication And Meta-analysis (DIAGRAM) consortium integrated data from three previously published GWAS (Saxena et al., 2007; Scott et al., 2007; Zeggini et al., 2007), thereby doubling the sample size compared to the largest of the individual studies to ~4500 cases and 5500 controls. The consortium also used novel imputation approaches (Marchini et al., 2007) to infer genotypes at additional SNPs that were not directly typed on the commercial arrays used for the original GWAS, thereby extending the analysis to a total of ~2.2 million SNPs across the genome. In this study, 69 signals showing the strongest associations in the GWAS meta-analysis were genotyped in an initial replication set of 22426 individuals and the top eleven signals emerging from this second analysis were then evaluated in ~57 000 further subjects. After integrating data from all study subjects, six signals reached combined levels of significance, including the JAZF1 (rs864745) (p = 5.0x10-14), CDC123-CAMK1D (rs12779790) (p = 1.2x1¯010), TSPAN8-LGR5 (rs7961581) (p = 1.1x10-9), THADA (rs7578597) (p = 1.1x10-9), ADAMTS9 (rs4607103) (p = 1.2x10-8) and NOTCH2 (rs10923931) (p = 4.1x10-8) (Zeggini et al., 2008). However, the replication study of these six SNPs in Khatri Sikh diabetics of North India, only CDC123/CAMKID (rs12779790) revealed a significant evidence of association with T2DM (pdominant = 0.031) (Sanghera et al., 2009).
NEW GENES NEW AETIOLOGY
The strongest signal for T2DM in GWAS was found for TCF7L2 with p-values down to <10-48 increase in the odds for the disease of 37% (Scott et al., 2007; Saxena et al., 2007), the TCF7L2 rs7901695, increases disease risk with an odds ratio of 1.37 (Table 1; Fig. 1). The expression of TCF7L2 was related to genotype and metabolic parameters in human islets, the risk T allele was associated with impaired insulin secretion, in cretin effects, and enhanced rate of hepatic glucose production. Over expression of TCF7L2 in human islets reduced glucose-stimulated insulin secretion (Lyssenko et al., 2007). The HHEX region which also harbors IDE encoding insulin-degrading enzyme which has been implicated in both insulin signal and islet function. T2DM risk allele for HHEX/IDE gene associated with decreased pancreatic β-cell function, including decreased β-cell glucose sensitivity that relates insulin secretion to plasma glucose concentration (Pascoe et al., 2007). Both HHEX and IDE are critical for ventral pancreas development and are powerful biological candidates for T2DM.
A central theme for many of the recently discovered genes is that many of them seem to be involved in insulin secretion, pin-pointing the pivotal role of β-cell function in the pathogenesis of T2DM. These genes include TCF7L2, KCNJ11, HHEX, SLC30A8, CDKAL1, CDKN2A/2B, IGF2BP2 and KCNQ1 (Steinthorsdottir et al., 2007; Grarup et al., 2007; Pascoe et al., 2007; Staiger et al., 2007, 2008; Yasuda et al., 2008; Unoki et al., 2008). The loci seem particularly to be associated with an increased risk of developing T2DM through a reduced insulin-secretory capacity. A further highly interesting finding was the fat mass and obesity associated FTO gene which predisposes to T2DM by altering Body Mass Index (BMI). It is expressed in hypothalamus (Frayling et al., 2007) which is the key brain region for influencing appetite. Interestingly, the GWAS have identified a separate set of SNPs that seem to represent the strongest common genetic risk factor for heart disease (myocardial infarction) (Zeggini et al., 2007; Helgadottir et al., 2007; McPherson et al., 2007). There is no correlation between the T2DM signal and the heart disease signal, but the latter does fall closer to the CDKN2 genes, which encodes p16INK4a. Over expression of p16INK4a results in a decreased islet proliferation and β-cell dysfunction in ageing mice (Krishnamurthy et al., 2006), the initial human physiology studies have not provided any evidence that the risk alleles alter insulin secretion, but the mouse phenotype strongly implicates β-cell dysfunction. CDKAL1 is highly expressed in human islets (Zeggini et al., 2007). CDKAL1 shares homology with the CDK5RAP1 gene, a known inhibitor of CDK5 activation. CDK5 is implicated in reduced β-cell function, through the formation of p35-CDK5 complexes, which down regulate insulin expression (Ubeda et al., 2006; Wei et al., 2005). T2DM risk allele for CDKAL1 gene associated with decreased pancreatic β-cell function, including decreased β-cell glucose sensitivity that relates insulin secretion to plasma glucose concentration (Pascoe et al., 2007). The IGF2BP2 binds to the key growth and insulin signaling molecule insulin-like growth factor 2 (IGFII) and is also expressed in the pancreatic islet (Zeggini et al., 2007). T2DM risk for CDKAL1, SLC30A8, IGF2BP2, and LOC387761 is specifically mediated through defects in insulin secretion (Palmer et al., 2008). The product of KCNQ1 gene can form hetero multimers with two other potassium channel proteins, KCNE1 and KCNE3. The SNP rs2237892 in KCNQ1 has known roles in cardiac muscle, it is not yet known how this gene could affect the risk for T2DM, but there is evidence that it is turned on in the pancreas (Yasuda et al., 2008).
Curiously, some of the recent studies suggest a possible explanation for previous much debated epidemiological observations that men with T2DM are less likely to develop prostate cancer. The same allele in HNF1B that predisposes to T2DM was protective of prostate cancer (Gudmundsson et al., 2007). Moreover, different variants in JAZF1 are associated with T2DM and with prostate cancer (Saxena et al., 2007; Zeggini et al., 2007; Scott et al., 2007; Thomas et al., 2008). In keeping with an effect on development and transcriptional processes these findings may not come as a surprise although the exact causal relationships remain to be investigated (Frayling et al., 2008).
Moreover, carriers of novel T2DM risk alleles within JAZF1, CDC123/CAMK1D, and TSPAN8 it’s suggested an impaired pancreatic β-cell function in glucose-tolerant regions in the cohort of middle-aged people (Grarup et al., 2008). CDC123 is regulated by nutrient availability in S. cerevisiae and has a role in cell cycle regulation. Taken together, evidence from GWAS implicating variants in or near CDKAL1, CDKN2A/B, CDC123 and CAMK1D suggests that cell cycle dysregulation may be a common pathogenetic mechanism in T2DM (Ridderstråle and Groop, 2009). Notch homologue 2, Drosophila (NOTCH2) is known to be involved in pancreatic development, but the mechanisms involved for the ADAMTS9 and THADA genes remain unclear.
MTNR1B is thought to participate in light-dependent functions in the retina and in melatonin’s neuronal regulation of circadian rhythmicity and sleep cycles. As certain sleep disorders, such as obstructive sleep apnea, result from obesity and are associated with insulin resistance (De Sousa et al., 2008; Kashyap and Defronzo, 2007). Very recently, three new studies identified and reported extremely strong, incontrovertible evidence that the MTNR1B is associated with high fasting glucose levels and increased risk of T2DM (Prokopenko et al., 2009; Bouatia-Naji et al., 2009; Lyssenko et al., 2009). MTNR1B could represent a new interesting candidate gene linking sleep disorders with T2DM. These new data implicate an association between the sleep-wake rhythm, the so-called circadian rhythm, and fasting glucose levels, T2DM, which was not known previously. The greatest benefit of T2DM genetic study is likely to come from new and better therapies derived from an improved understanding of the aetiology of the disease.
PREDICTION OF RISK OF T2DM
One of objectives for identification of T2DM susceptibility genes is to predict T2DM risk and identify high-risk subjects. To predict the risk of T2DM for a healthy individual we need to know and be able to measure risk factors, their effect sizes and how they interact. Although, prediction of total risk is an ultimate goal, prediction of genetic risk that can be attributed to inherited genetic variants is an important component. Generally, the predictive value of genetic testing for prediction of T2DM is unclear. Several empirical studies on the predictive value of genetic polymorphisms have been conducted before the GWAS data were available (Lyssenko et al., 2005; Weedon et al., 2006). In a case-control study, combining the information of three polymorphisms improved disease prediction, albeit to a limited extent (Weedon et al., 2006). The predictive value was low compared with clinical characteristics (Vaxillaire et al., 2008). The complex disease is caused by multiple genetic variants, the predictive testing based on a single genetic marker will be of limited value. GWAS have dramatically increased the number of common genetic variants that are robustly associated with T2DM. The predictive value could be improved by combining multiple common low-risk variants, but all showed limited predictive value so far (Janssens et al., 2006, 2007; Yang et al., 2003; Wray et al., 2007). The currently known and replicated genetic variants found in GWAS contributed modestly to the prediction of T2DM in population-based setting and marginally improved the risk prediction beyond clinical characteristics (Van Hoek et al., 2008). Individuals carrying more risk alleles had a higher risk of T2DM and the common risk variants for T2DM do not provide strong predictive value at a population level (Lango et al., 2008). Similarly Lin et al. (2009) constructed an additive genetic score using the most replicated SNPs within 15 T2DM susceptibility genes and reported that the weighted 15 SNP-based genetic score provides additional information over clinical predictors of prevalent T2DM. However, the clinical benefit of this genetic information is limited. The prediction model for T2DM may not be so useful but has some value and incorporation of data from additional risk loci is most likely to increase the predictive power (Miyake et al., 2009). New gene discoveries from GWAS will certainly identify novel etiological pathways and novel intermediate biomarkers, which consequently may be stronger predictors of disease than the genetic variant that led to its identification. But it is likely that the complexity of complex diseases may ultimately limit the opportunities for accurate prediction of disease in asymptomatic individuals as unraveling their complete causal pathways may be impossible (Janssens and van Duijn, 2008). More extensive studies are needed to assess the usefulness of combining information from multiple variants.
CONCLUSION
Studies in human genetics have made tremendous strides in discovering T2DM genes especially through GWAS. Identification of the causal variants responsible for the association signals uncovered will provide valuable clues to understanding disease predisposition. GWAS is only the beginning in establishing the role of genetic variants in disease etiology. This would be followed by the fine-mapping of the susceptibility region through deep sequencing in large population and the validation of causality for genetic variants in experimental settings.
| • | Fine-map the new T2DM gene regions. That will involve deep sequencing and further rounds of genotyping to build up a full picture of all the possible common variation that might explain the association signals. This should include efforts to define Copy Number Variants (CNVs) such as duplications and deletions, and should also attempt to define independent associations in the same gene regions |
| • | Additional association studies of the new variants are needed. Investigators will need to assess their role in other populations, especially populations with a high prevalence of T2DM. Further studies of the role of risk alleles in the general population are also important |
| • | The predictive value of genetic testing for prediction of T2DM is unclear. Additional studies are needed to identify and replicate new genetic susceptibility variants and gene-gene and gene-environment interactions to approach levels of discriminative accuracy that enable the identification of at-risk groups and to assess whether individuals with extreme numbers of risk alleles may benefit from genetic testing |
| • | The mechanisms whereby a given DNA change leads to an increased risk of T2DM need to be reconstructed. We need to know whether they influence T2DM predisposition through primary effects on β-cell function, through insulin action, or by some other mechanism. The ultimate objective is, of course, to understand how genetic findings can translate into advances in clinical management. In principle, genetic testing might offer insights into disease risk and predict response to the various therapeutic and preventative options available, but much work will be required to understand how to deploy such tests in clinically effective ways. A route is to apply the insights gained into the mechanisms of disease predisposition to identify new targets for drug development. In this respect, genes of small effect are likely to provide clues just as valuable as those of large effect |
Looking ahead, new methodologies and approaches may be needed to discover the remaining as yet unidentified genetic contributors to disease risk. At associated loci, fine mapping can help narrow down the list of possible causal variants and simplify future functional studies. Additional GWAS, in larger samples and multiple ethnicities, will almost certainly lead to new discoveries and incremental gains in the amount of risk accounted for by identified genetic variants. Exploration of these novel loci will very likely uncover additional alleles, both common and rare, that explain additional variance in phenotype, help pinpoint which gene(s) are responsible for the association and provide better clinical and molecular tools for assessing function and mechanism of disease.
REFERENCES
- Abate, N., M. Chandalia, P. Satija, B. Adams-Huet and S.M. Grundy et al., 2001. ENPP1/PC-1 K121Q polymorphism and genetic susceptibility to type 2 diabetes. Diabetes, 54: 1207-1213.
Direct Link - Altshuler, D., J.N. Hirschhorn, M. Klannemark, C.M. Lindgren and M.C. Vohl et al., 2000. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet., 26: 76-80.
PubMedDirect Link - Arfa, I., A. Abid, D. Malouche, N. Ben Alaya and T.R. Azegue et al., 2007. Familial aggregation and excess maternal transmission of type 2 diabetes in Tunisia. Postgrad. Med. J., 83: 348-351.
CrossRefPubMedDirect Link - Aulchenko, Y.S., J. Pullen, W.P. Kloosterman, M. Yazdanpanah and A Hofman et al., 2007. LPIN2 is associated with type 2 diabetes, glucose metabolism and body composition. Diabetes, 56: 3020-3026.
PubMedDirect Link - Babenko, A.P., M. Polak, H. Cave, K. Busiah and P. Czernichow et al., 2006. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med., 355: 456-466.
PubMedDirect Link - Bacci, S., O. Ludovico, S. Prudente, Y.Y. Zhang and R. Di Paola et al., 2005. The K121Q polymorphism of the ENPP1/PC-1 gene is associated with insulin resistance/atherogenic phenotypes, including earlier onset of type 2 diabetes and myocardial infarction. Diabetes, 54: 3021-3025.
Direct Link - Baier, L.J., P.A. Permana, X. Yang, R.E. Pratley and R.L. Hanson et al., 2000. A calpain-10 gene polymorphism is associated with reduced muscle mRNA levels and insulin resistance. J. Clin. Invest., 106: R69-R73.
PubMedDirect Link - Barroso, I., J. Luan, R.P. Middelberg, A.H. Harding and P.W. Franks et al., 2003. Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS. Biol., 1: e20-e20.
PubMedDirect Link - Bento, J.L., N.D. Palmer, J.C. Mychaleckyj, L.A. Lange and C.D. Langefeld et al., 2004. Association of protein tyrosine phosphatase 1B gene polymorphisms with type 2 diabetes. Diabetes, 53: 3007-3012.
PubMedDirect Link - Bingham, C., S. Ellard, L. Allen, M. Bulman and M. Shepherd et al., 2000. Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1 beta. Kidney Int., 57: 898-907.
CrossRefPubMedDirect Link - Bonnycastle, L.L., C.J. Willer, K.N. Conneely, A.U. Jackson and C.P. Burrill et al., 2006. Common variants in maturity-onset diabetes of the young genes contribute to risk of type 2 diabetes in Finns. Diabetes, 55: 2534-2540.
CrossRefPubMedDirect Link - Bouatia-Naji, N., A. Bonnefond, C. Cavalcanti-Proen�a, T. Spars� and J. Holmkvist et al., 2009. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet., 41: 89-94.
PubMedDirect Link - Chang, Y.C., T.J. Chang, Y.D. Jiang, S.S. Kuo, K.C. Lee, K.C. Chiu and L.M. Chuang, 2007. Association study of the genetic polymorphisms of the transcription factor 7-like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes, 56: 2631-2637.
CrossRefPubMedDirect Link - Cauchi, S., D. Meyre, C. Dina, H. Choquet and C. Samson et al., 2006. Transcription factor TCF7L2 genetic study in the French population: Expression in human β-cells and adipose tissue and strong association with type 2 diabetes. Diabetes, 55: 2903-2908.
CrossRefPubMedDirect Link - Cauchi, S., Y. El Achhab, H. Choquet, C. Dina and F. Krempler et al., 2007. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: A global meta-analysis. J. Mol. Med., 85: 777-782.
CrossRefPubMedDirect Link - Damcott, C.M., T.I. Pollin, L.J. Reinhart, S.H. Ott and H. Shen et al., 2006. Polymorphisms in the Transcription Factor 7-Like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: Replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes, 55: 2654-2659.
CrossRefPubMedDirect Link - Deeb, S.S., L. Fajas, M. Nemoto, J. Pihlajam�ki and L. Mykk�nen et al., 1998. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet., 20: 284-287.
Direct Link - Dehwah, M.A.S., Z. Shuang, W. Yan, P. Chan and Q.Y. Huang, 2008. Type 2 diabetes in han chinese in hubei. J. Applied Sci., 8: 1235-1241.
CrossRefDirect Link - Demenais, F., T. Kanninen, C.M. Lindgren, S. Wiltshire and S. Gaget et al., 2003. A meta-analysis of four European genome screens (GIFT Consortium) shows evidence for a novel region on chromosome 17p11.2-q22 linked to type 2 diabetes. Hum. Mol. Genet., 12: 1865-1873.
CrossRefPubMedDirect Link - De Silva, S.N., N. Weerasuriya, N.M. De Alwis, M.W. de Silva and D.J. Fernando, 2002. Excess maternal transmission and familial aggregation of type 2 diabetes in Sri Lanka. Diabetes Res. Clin. Pract., 58: 173-177.
PubMedDirect Link - De Sousa, A.G., C. Cercato, M.C. Mancini and A. Halpern, 2008. Obesity and obstructive sleep apnea-hypopnea syndrome. Obesity Rev., 9: 340-354.
CrossRefPubMedDirect Link - Dina, C., D. Meyre, S. Gallina, E. Durand and A. Korner et al., 2007. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet., 39: 724-726.
CrossRefPubMedDirect Link - Elbein, S.C., J. Sun, E. Scroggin, K. Teng and S.J. Hasstedt, 2001. Role of common sequence variants in insulin secretion in familial type 2 diabetic kindreds: the sulfonylurea receptor, glucokinase, and hepatocyte nuclear factor 1alpha genes. Diabetes Care, 24: 472-478.
CrossRefPubMedDirect Link - Fajans, S.S., G.I. Bell and K.S. Polonsky, 2001. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med., 345: 971-980.
PubMedDirect Link - Florez, J.C., N. Burtt, P.I. de Bakker, P. Almgren and T. Tuomi et al., 2004. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes, 53: 1360-1368.
CrossRefPubMedDirect Link - Florez, J.C., C.M. Agapakis, N.P. Burtt, M. Sun and P. Almgren et al., 2005. Association testing of the protein tyrosine phosphatase 1B gene (PTPN1) with type 2 diabetes in 7, 883 people. Diabetes, 54: 1884-1891.
PubMedDirect Link - Florez, J.C., A.K. Manning, J. Dupuis, J. McAteer and K. Irenze et al., 2007. A 100k genome-wide association scan for diabetes and related traits in the Framingham Heart Study: Replication and integration with other genome-wide datasets. Diabetes, 56: 3063-3074.
CrossRefPubMedDirect Link - Franks, P.W., O. Rolandsson, S.L. Debenham, K.A. Fawcett and F. Payne et al., 2008. Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia, 51: 458-463.
CrossRefPubMedDirect Link - Frayling, T.M., N.J. Timpson, M.N. Weedon, E. Zeggini and R.M. Freathy et al., 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science, 316: 889-894.
CrossRefPubMedDirect Link - Frayling, T.M., 2007. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat. Rev. Genet., 8: 657-662.
PubMedDirect Link - Frayling, T.M., H. Colhoun and J.C. Florez, 2008. A genetic link between type 2 diabetes and prostate cancer. Diabetologia, 51: 1757-1760.
CrossRefPubMedDirect Link - Gibson, F. and P. Froguel, 2004. Genetics of the APM1 locus and its contribution to type 2 diabetes susceptibility in French Caucasians. Diabetes, 53: 2977-2983.
CrossRefPubMedDirect Link - Gloyn, A.L., M.N. Weedon, K.R. Owen, M.J. Turner and B.A. Knight et al., 2003. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes, 52: 568-572.
CrossRefPubMedDirect Link - Gloyn, A.L., E.R. Pearson, J.F. Antcliff, P. Proks and G.J. Bruining et al., 2004. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med., 350: 1838-1849.
PubMedDirect Link - Grant, S.F., G. Thorleifsson, I. Reynisdottir, R. Benediktsson and A. Manolescu et al., 2006. Variant of Transcription Factor 7-Like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet., 38: 320-323.
CrossRefPubMedDirect Link - Grarup, N., C.S. Rose, E.A. Andersson, G. Andersen and A.L. Nielsen et al., 2007. Studies of association of variants near the HHEX, CDKN2A/B and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: Validation and extension of genome-wide association studies. Diabetes, 56: 3105-3111.
CrossRefPubMedDirect Link - Grarup, N., G. Andersen, N.T. Krarup, A. Albrechtsen and O. Schmitz et al., 2008. Association testing of novel type 2 diabetes risk alleles in the JAZF1, CDC123/CAMK1D, TSPAN8, THADA, ADAMTS9 and NOTCH2 loci with insulin release, insulin sensitivity and obesity in a population-based sample of 4,516 glucose-tolerant middle-aged Danes. Diabetes, 57: 2534-2540.
CrossRefPubMedDirect Link - Groves, C.J., E. Zeggini, J. Minton, T.M. Frayling and M.N. Weedon et al., 2006. Association analysis of 6736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes, 55: 2640-2644.
PubMedDirect Link - Gudmundsson, J., P. Sulem, V. Steinthorsdottir, J.T. Bergthorsson and G. Thorleifsson et al., 2007. Two variants on chromosome 17 confer prostate cancer risk and the one in TCF2 protects against type 2 diabetes. Nat. Genet., 39: 977-983.
PubMedDirect Link - Hansen, T., S.M. Echwald, L. Hansen, A.M. M�ller and K. Almind et al., 1998. Decreased tolbutamide-stimulated insulin secretion in healthy subjects with sequence variants in the high-affinity sulfonylurea receptor gene. Diabetes, 47: 598-605.
Direct Link - Hani, E.H., J. Hager, A. Philippi, F. Demenais, P. Froguel and N. Vionnet, 1997. Mapping NIDDM susceptibility loci in French families: Studies with markers in the region of NIDDM1 on chromosome 2q. Diabetes, 46: 1225-1226.
PubMedDirect Link - Hanson, R.L., C. Bogardus, D. Duggan, S. Kobes and M. Knowlton et al., 2007. A search for variants associated with young-onset type 2 diabetes in American Indians in a 100k genotyping array. Diabetes, 56: 3045-3052.
CrossRefPubMedDirect Link - Hart, L.M., P. de Knijff, J.M. Dekker, R.P. Stolk and G. Nijpels et al., 1999. Variants in the sulphonylurea receptor gene: Association of the exon 16-3t variant with type II diabetes mellitus in Dutch Caucasians. Diabetologia, 42: 617-620.
PubMedDirect Link - Hayes, M.G., A. Pluzhnikov, K. Miyake, Y. Sun and M.C.Y. Ng et al., 2007. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes, 56: 3033-3044.
Direct Link - Hegele, R.A., F. Sun, S.B. Harris, C. Anderson, A.J. Hanley and B. Zinman, 1999. Genome-wide scanning for type 2 diabetes susceptibility in Canadian Oji-Cree, using 190 microsatellite markers. J. Hum. Genet., 44: 10-14.
Direct Link - Helgadottir, A., G. Thorleifsson, A. Manolescu, S. Gretarsdottir and T. Blondal et al., 2007. A common variant on chromosome 9p21 affects risk of myocardial infarction. Science, 316: 1491-1493.
CrossRefPubMedDirect Link - Herder, C., W. Rathmann, K. Strassburger, H. Finner and H. Grallert et al., 2008. Variants of the PPARG, IGF2BP2, CDKAL1, HHEX and TCF7L2 genes confer risk of type 2 diabetes independently of BMI in the German KORA studies. Horm. Metab. Res., 40: 722-726.
PubMedDirect Link - Holmkvist, J., P. Almgren, V. Lyssenko, C.M. Lindgren and K.F. Eriksson et al., 2008. Common variants in maturity-onset diabetes of the young genes and future risk of type 2 diabetes. Diabetes, 57: 1738-1744.
CrossRefPubMedDirect Link - Horikawa, Y., N. Oda, N.J. Cox, X. Li and M. Orho-Melander et al., 2000. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet., 26: 163-175.
Direct Link - Hu, C., C. Wang, R. Zhang, X. Ma and J. Wang et al., 2009. Variations in KCNQ1 are associated with type 2 diabetes and beta cell function in a Chinese population. Diabetologia, 52: 1322-1325.
CrossRefPubMedDirect Link - Huang, Q.Y., M.R. Cheng and S.L. Ji, 2006. Linkage and association studies of the susceptibility genes for type 2 diabetes. Acta Genet. Sin., 33: 573-589.
CrossRefDirect Link - Inoue, H., J. Ferrer, C.M. Welling, S.C. Elbein and M. Hoffman et al., 1996. Sequence variants in the sulfonylurea receptor (SUR) gene are associated with NIDDM in Caucasians. Diabetes, 45: 825-831.
CrossRefPubMedDirect Link - Inoue, H., J. Ferrer, M. Warren-Perry, Y. Zhang and H. Millns et al., 1997. Sequence variants in the pancreatic islet beta-cell inwardly rectifying K+ channel Kir6.2 (Bir) gene: identification and lack of role in Caucasian patients with NIDDM. Diabetes, 46: 502-507.
Direct Link - Janssens, A.C., Y.S. Aulchenko, S. Elefante, G.J. Borsboom, E.W. Steyerberg and C.M. van Duijn, 2006. Predictive testing for complex diseases using multiple genes: Factor or fiction? Genet. Med., 8: 395-400.
PubMedDirect Link - Janssens, A.C., R. Moonesinghe, Q. Yang, E.W. Steyerberg, C.M. van Duijn and M.J. Khoury, 2007. The impact of genotype frequencies on the clinical validity of genomic profiling for predicting common chronic diseases. Genet. Med., 9: 528-535.
PubMedDirect Link - Janssens, A.C.J.W. and C.M. van Duijn, 2008. Genome-based prediction of common diseases: Advances and prospects. Hum. Mol. Genet., 17: R166-R173.
CrossRefPubMedDirect Link - Kashyap, S.R. and R.A. DeFronzo, 2007. The insulin resistance syndrome: Physiological considerations. Diab. Vasc. Dis. Res., 4: 13-19.
CrossRefPubMedDirect Link - King, H. and M. Rewers, 1993. Global estimates for prevalence of diabetes mellitus and impaired glucose tolerance in adults. WHO Ad Hoc diabetes reporting group. Diabetes Care, 16: 157-177.
CrossRefPubMedDirect Link - Kissebah, A.H., G.E. Sonnenberg, J. Myklebust, M. Goldstein and K. Broman et al., 2000. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc. Natl. Acad. Sci. USA., 97: 14478-14483.
PubMedDirect Link - Knowler, W.C., R.C. Williams, D.J. Pettitt and A.G. Steinberg, 1988. Gm3: 5,13,14 and type 2 diabetes mellitus: An association in American Indians with genetic admixture. Am. J. Hum. Genet., 43: 520-526.
PubMedDirect Link - Krishnamurthy, J., M.R. Ramsey, K.L. Ligon, C. Torrice, A. Koh, S. Bonner-Weir and N.E. Sharpless, 2006. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature, 443: 453-457.
PubMedDirect Link - Lango, H., C.N.A. Palmer, A.D. Morris, E. Zeggini and A.T. Hattersley et al., 2008. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes, 57: 3129-3135.
CrossRefPubMedDirect Link - Lee, Y.H., E.S. Kang, S.H. Kim, S.J. Han and C.H. Kim et al., 2008. Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J. Hum. Genet., 53: 991-998.
CrossRefPubMedDirect Link - Li, S., L. Li, K. Li, X. Qi, D. Hoekema, H. Liu and G. Yang, 2008. Association of adipose most abundant transcript 1 gene (apM1) with type 2 diabetes mellitus in a Chinese population: A meta-analysis of case-control studies. Clin. Endocrinol. (Oxf)., 68: 885-889.
PubMedDirect Link - Lin, X., K. Song, N. Lim, X. Yuan and T. Johnson et al., 2009. Risk prediction of prevalent diabetes in a Swiss population using a weighted genetic score-the CoLaus Study. Diabetologia, 52: 600-608.
CrossRefPubMedDirect Link - Lindner, T.H., P.R. Njolstad, Y. Horikawa, L. Bostad, G.I. Bell and O. Sovik, 1999. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet., 8: 2001-2008.
Direct Link - Liu, Y., L. Yu, D. Zhang, Z. Chen and D.Z. Zhou et al., 2008. Positive association between variations in CDKAL1 and type 2 diabetes in Han Chinese individuals. Diabetologia, 51: 2134-2137.
CrossRefPubMedDirect Link - Liu, Y., D.Z. Zhou, D. Zhang, Z. Chen and T. Zhao et al., 2009. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes in the population of mainland China. Diabetologia, 52: 1315-1321.
CrossRefPubMedDirect Link - Love-Gregory, L., J. Wasson, J. Lin, G. Skolnick, B. Suarez and M.A. Permutt, 2003. E23K single nucleotide polymorphism in the islet ATP-sensitive potassium channel gene (Kir6.2) contributes as much to the risk of type II diabetes in Caucasians as the PPARgamma Pro12Ala variant. Diabetologia, 46: 136-137.
CrossRefPubMedDirect Link - Love-Gregory, L.D., J. Wasson, J. Ma, C.H. Jin, B. Glaser, B.K. Suarez and M.A. Permutt, 2004. A common polymorphism in the upstream promoter region of the hepatocyte nuclear factor-4 alpha gene on chromosome 20q is associated with type 2 diabetes and appears to contribute to the evidence for linkage in an ashkenazi jewish population. Diabetes, 53: 1134-1140.
CrossRefPubMedDirect Link - Lyssenko, V., P. Almgren, D. Anevski, M. Orho-Melander and M. Sjogren et al., 2005. Genetic prediction of future type 2 diabetes. PLOS Med., 2: e345-e345.
Direct Link - Lyssenko, V., R. Lupi, P. Marchetti, S. Del Guerra and M. Orho-Melander et al., 2007. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest., 117: 2155-2163.
CrossRefPubMedDirect Link - Lyssenko, V., A. Jonsson, P. Almgren, N. Pulizzi and B. Isomaa et al., 2008. Clinical risk factors, DNA variants and the development of type 2 diabetes. N. Engl. J. Med., 359: 2220-2232.
PubMedDirect Link - Lyssenko, V., C.L. Nagorny, M.R. Erdos, N. Wierup and A. Jonsson et al., 2009. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet., 41: 82-88.
PubMedDirect Link - Marchini, J., B. Howie, S. Myers, G. McVean and P. Donnelly, 2007. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet., 39: 906-913.
Direct Link - Matsuoka, N., A. Patki, H.K. Tiwari, D.B. Allison and S.B. Johnson et al., 2006. Association of K121Q polymorphism in ENPP1 (PC-1) with BMI in Caucasian and African-American adults. Int. J. Obesity (Lond.), 30: 233-237.
Direct Link - McPherson, R., A. Pertsemlidis, N. Kavaslar, A. Stewart and R. Roberts et al., 2007. A common allele on chromosome 9 associated with coronary heart disease. Science, 316: 1488-1491.
CrossRefDirect Link - Meirhaeghe, A., N. Helbecque, D. Cottel, D. Arveiler and J.B. Ruidavets et al., 2001. Impact of sulfonylurea receptor 1 genetic variability on non-insulin-dependent diabetes mellitus prevalence and treatment: A population study. Am. J. Med. Genet., 101: 4-8.
PubMedDirect Link - Minton, J.A.L., A.T. Hattersley, K. Owen, M.I. McCarthy and M. Walker et al., 2002. Association studies of genetic variation in the WFS1 gene and type 2 diabetes in UK populations. Diabetes, 51: 1287-1290.
CrossRefPubMedDirect Link - Ng, M.C.Y., C.H.T. Tam, V.K.L. Lam, W.Y. So, R.C.W. Ma and J.C.N. Chan, 2007. Replication and identification of novel variants at TCF7L2 associated with type 2 diabetes in Hong Kong Chinese. J. Clin. Endocrinol. Metab., 92: 3733-3737.
CrossRefPubMedDirect Link - Palmer, N.D., J.L. Bento, J.C. Mychaleckyj, C.D. Langefeld and J.K. Campbell et al., 2004. Association of protein tyrosine phosphatase 1B gene polymorphisms with measures of glucose homeostasis in Hispanic Americans: The insulin resistance atherosclerosis study (IRAS) family study. Diabetes, 53: 3013-3019.
CrossRefPubMed - Rampersaud, E., C.M. Damcott, M. Fu, H. Shen and P. McArdle et al., 2007. Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the old order Amish: Evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes, 56: 3053-3062.
CrossRefPubMed - Reppert, S.M., C. Godson, C.D. Mahle, D.R. Weaver, S.A. Slaugenhaupt and J.F. Gusella, 1995. Molecular characterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA., 92: 8734-8738.
Direct Link - Rong, R., R.L. Hanson, D. Ortiz, C. Wiedrich, S. Kobes, W.C. Knowler, C. Bogardus and L.J. Baier, 2009. Association analysis of variation in/near FTO, CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2, LOC387761 and CDKN2B with type 2 diabetes and related quantitative traits in Pima Indians. Diabetes, 58: 478-488.
CrossRefPubMed - Sakura, H., N. Wat, V. Horton, H. Millns, R.C. Turner and F.M. Ashcroft, 1996. Sequence variations in the human Kir6.2 gene, a subunit of the beta- cell ATP-sensitive K-channel: No association with NIDDM in while Caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia, 39: 1233-1236.
CrossRefPubMed - Saxena, R., B.F. Voight , V. Lyssenko , N.P. Burtt and P.I. de Bakker et al., 2007. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science, 316: 1331-1336.
CrossRefPubMedDirect Link - Scott, L.J., L.L. Bonnycastle, C.J. Willer, A.G. Sprau and A.U. Jackson et al., 2006. Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes, 55: 2649-2653.
CrossRefPubMedDirect Link - Schwanstecher, C., U. Meyer and M. Schwanstecher, 2002. K (IR) 6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta-cell ATP-sensitive K (+) channels. Diabetes, 51: 875-879.
CrossRefPubMedDirect Link - Scott, L.J., K.L. Mohlke, L.L. Bonnycastle, C.J. Willer, Y. Li and W.L. Duren et al., 2007. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science, 316: 1341-1345.
CrossRefPubMedDirect Link - Serjeantson, S.W., D. Owerbach, P. Zimmet, J. Nerup and K. Thoma, 1983. Genetics of diabetes in Nauru: Effects of foreign admixture, HLA antigens and the insulin-gene linked polymorphism. Diabetologia, 25: 13-17.
CrossRefPubMedDirect Link - Shimomura, I., R.E. Hammer, S. Ikemoto, M.S. Brown and J.L. Goldstein, 1999. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature, 401: 73-76.
CrossRefPubMedDirect Link - Silander, K., K.L. Mohlke, L.J. Scott, E.C. Peck and P. Hollstein et al., 2004. Genetic variation near the hepatocyte nuclear factor-4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes, 53: 1141-1149.
CrossRefPubMedDirect Link - Sladek, R., G. Rocheleau, J. Rung, C. Dina and L. Shen et al., 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445: 881-885.
CrossRefPubMedDirect Link - Staiger, H., F. Machicao, N. Stefan, O. Tschritter and C. Thamer et al., 2007. Polymorphisms within novel risk loci for type 2 diabetes determine beta-cell function. PLoS O. N. E., 2: e832-e832.
CrossRefPubMedDirect Link - Staiger, H., A. Stancakova, J. Zilinskaite, M. V�nttinen and T. Hansen et al., 2008. A candidate type 2 diabetes polymorphism near the HHEX locus affects acute glucose-stimulated insulin release in European populations: results from the EUGENE2 study. Diabetes, 57: 514-517.
CrossRefPubMedDirect Link - Steinthorsdottir, V., G. Thorleifsson, I. Reynisdottir, R. Benediktsson and T. Jonsdottir et al., 2007. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet., 39: 770-775.
CrossRefPubMedDirect Link - Tabara, Y., H. Osawa, R. Kawamoto, H. Onuma and I. Shimizu et al., 2009. Replication study of candidate genes associated with type 2 diabetes based on genome-wide screening. Diabetes, 58: 493-498.
CrossRefPubMedDirect Link - Thomas, F., B. Balkau, F. Vauzelle-Kervroedan and L. Papoz, 1994. Maternal effect and familial aggregation in NIDDM. The CODIAB study. CODIAB-INSERM-ZENECA study group. Diabetes, 43: 63-67.
CrossRefPubMedDirect Link - Thomas, G., K.B. Jacobs, M. Yeager, P. Kraft, S. Wacholder et al., 2008. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet., 40: 310-315.
CrossRefPubMedDirect Link - Tong, Y., Y. Lin, Y. Zhang, J. Yang, Y. Zhang, H. Liu and B. Zhang, 2009. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large human genome epidemiology (HuGE) review and meta-analysis. BMC Med. Genet., 10: 15-15.
CrossRefPubMedDirect Link - Ubeda, M., J.M. Rukstalis and J.F. Habener, 2006. Inhibition of cyclin-dependent kinase 5 activity protects pancreatic beta cells from glucotoxicity. J. Biol. Chem., 281: 28858-28864.
CrossRefPubMedDirect Link - Van Tilburg, J.H., L.A. Sandkuijl, E. Strengman, H.V. Someren and C. Wijmenga et al., 2003. A genome-wide scan in type 2 diabetes mellitus provides independent replication of a susceptibility locus on 18p11 and suggests the existence of novel Loci on 2q12 and 19q13. J. Clin. Endocrinol. Metab., 88: 2223-2230.
CrossRefPubMed - Vionnet, N., E.H. Hani, S. Dupont, S. Gallina and S. Francke et al., 2000. Genomewide search for type 2 diabetes susceptibility genes in French whites evidence for a novel susceptibility locus for early onset diabetes on chromosome 3q27 qter and independent replication of a type 2 diabetes locus on chromosome 1q21 q24. Am. J. Hum. Genet., 67: 1470-1480.
CrossRefPubMed - Weedon, M.N., M.I. McCarthy, G. Hitman, M. Walker and C.J. Groves et al., 2006. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLOS Med., Vol. 3, No. 10.
CrossRefDirect Link - Zacharova, J., J.L. Chiasson, M. Laakso and S.N. Study Group, 2005. The common polymorphisms (single nucleotide polymorphism [SNP] +45 and SNP +276) of the adiponectin gene predict the conversion from impaired glucose tolerance to type 2 diabetes: the STOP-NIDDM trial. Diabetes, 54: 893-899.
CrossRefPubMed - Zeggini, E., M.W. Weedon, C.M. Lindgren, T.M. Frayling and K.S. Elliott et al., 2007. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science, 316: 1336-1341.
CrossRefPubMedDirect Link - Zouali, H., E.H. Hani, A. Philippi, N. Vionnet, J.S. Beckmann, F. Demenais and P. Froguel, 1997. A susceptibility locus for early-onset non-insulin dependent (type 2) diabetes mellitus maps to chromosome 20q, proximal to the phosphoenolpyruvate carboxykinase gene. Hum. Mol. Genet., 6: 1401-1408.
PubMedDirect Link