
Anusha Bhaskar
PRIST University, Vallam, Thanjavur 613 403, India
V. Nithya
Srimad Andavan College, Trichy, India
Trends in Bioinformatics
Year: 2013 | Volume: 6 | Issue: 2 | Page No.: 45-61







ABSTRACT
Keratitis is a condition in which the cornea of the eye becomes inflamed. It is caused by bacteria, viruses, fungi and parasites. The diagnosis of the causative organism of keratitis remains a problem due to lack of advanced techniques. In this study PCR and sequence analysis of 16S rDNA was used to ascertain the bacteria isolated in eye swabs of keratitis patients. The bacterial genes were sequenced and deposited in the GenBank (NCBI) with the accession numbers JN378393, JN378392, HM204502, JQ039348, JN652127, JN652129, JQ039350, JN652128, JQ039349 and HQ404365 for Streptococcus viridan, Staphylococcus aureus, Micrococcus sp., Moraxella sp., Citrobacter koseri, Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella sp., Propionibacterium sp. and Staphylococcus epidermidis, respectively. Sequences were analyzed and aligned by ClustalX and a phylogenetic relationship was studied. The secondary structure of the 16S DNA was developed using GeneBee which produces secondary structures prediction through the minimization of the potential energy of the system. These results may be useful in characterizing the micro evolutionary mechanisms of the species for researchers.
PDF Abstract XML References Citation
Received: September 12, 2012;
Accepted: April 01, 2013;
Published: June 14, 2013
How to cite this article
Anusha Bhaskar and V. Nithya, 2013. In silico Structural Analysis of 16S rDNA Sequences of Bacteria Isolated from Keratitis Patients. Trends in Bioinformatics, 6: 45-61.
DOI: 10.3923/tb.2013.45.61
URL: https://scialert.net/abstract/?doi=tb.2013.45.61
DOI: 10.3923/tb.2013.45.61
URL: https://scialert.net/abstract/?doi=tb.2013.45.61
INTRODUCTION
Microbial keratitis is a leading cause of visual loss in developing countries. It is normally predisposed by ocular trauma and/or contact lens wear. Corneal scarring listed second only to cataracts as an important cause of blindness and visual impairment in many developing countries in Asia, Africa and the Middle East (Whitcher and Srinivasan, 1997). Bacterial keratitis is the most common form of suppurative corneal ulceration. A number of organisms are able to cause infection (Dunlop et al., 1994; Upadhyay et al., 1991; Wilhelmus, 1996) and analyses of them are essential to understand how they set up the infection.
Approximately 65-90% of all corneal infections is bacterial keratitis (Asbell and Stenson, 1982; McDonnell and Green, 1990; Jones, 1981; Laibson, 1972; Liesegang and Forster, 1980). Although, these bacteria may vary in incidence according to geographical locale (Baum, 1979) the most common organisms include Staphylococcus aureus, Streptococcus epidermis, Streptococcus pneumoniae, Pseudomonas aeruginosa and other Gram negative bacilli (Morlet and Daniell, 2003; Jones, 1979; Baum, 1978). Of these, S. aureus is the predominant pathogen isolated from the majority of cases of keratitis, (Morlet and Daniell, 2003; Baum, 1978) but P. aeruginosa, a potentially devastating ocular pathogen, is the most common cause of hypopyon corneal ulcers, (Hyndiuk, 1981) ulcerative keratitis associated with contact lens wear and severe necrotic corneal ulceration (Laibson, 1972).
In this study, 16S rDNA sequences of bacteria isolated from infectious keratitis in eye. Clinic of the General Hospital of Tiruchirappalli Tamil Nadu were sequenced. A comparative phylogenetic analysis of the 16S rDNA sequences were performed by using ClustalX and the phylogenetic tree was constructed using NJ method. The secondary structure of the 16S rDNA was constructed and minimal free energy was calculated by Genebee tool. The ribosomal RNA mainly 16S r DNA has proven to be a stable and specific molecular marker of the identification of bacteria. The 16S rDNA is a common target for the taxonomical purpose, mainly due to the mosaic composition of phylogenetically conserved and variable regions.
MATERIALS AND METHODS
Sample collection: In this study, bacteria were isolated from keratitis patients in Eye Clinic of the General Hospital of Tiruchirappalli Tamil Nadu during 2010-2012 All patients were examined under a slit-lamp biomicroscope by an ophthalmologist. Corneal scrapings were collected after instillation of 4% lignocaine without preservative under aseptic conditions from each ulcer by an ophthalmologist using a sterile Bard Parker blade (No. 15). Scrapings were performed under magnification of slit-lamp or operating microscope. Leading edge and base of each ulcer were scraped initially and the material obtained were directly inoculated onto the surface of solid media such as sheep blood agar, chocolate agar and Sabouraud Dextrose Agar (SDA) in a row of C-shaped streaks and also deep inoculation in the liquid media such as Brain Heart Infusion (BHI) broth without gentamicin sulphate and thioglycollate medium. Subsequent scrapings were spread onto labelled slides in a thin, even manner for 10% potassium hydroxide (KOH) wet mount and Gram staining.
Culture: All bacterial cultures were incubated aerobically. Cultures on blood agar and chocolate agar were evaluated at 24 hours and at 48 hours and then discarded if no growth was seen. All media’s were incubated at 35°C (±1) except SDA which are incubated at 27°C (±1) in BOD incubator. Cultures inoculated in BHI broth were examined for turbidity in similar fashion which was subsequently subcultured and Gram stained for identification. However, liquid media were prone to contamination and were not used for interpretation in isolation. The criteria described by Bharathi et al. (2002) were used for determining culture positive samples.
The specific identification of bacterial pathogens was based on microscopic morphology, staining characteristics and biochemical properties using standard laboratory criteria.
PCR and DNA sequencing: Genomic DNA was isolated from sample and loaded on 1% agarose gel. PCR amplified was performed using 16s rDNA primers (PCR amplification gel Photo attached). The PCR product was gel eluted. The purified PCR product was sequenced using 2 primers. The conditions used for PCR amplification of DNA: 1, 16S Forward Primer 400 ng, 16S Reverse Primer 400 ng, dNTPs (2.5 mM each) 4, 10X Taq DNA Polymerase Assay Buffer 10, Taq DNA Polymerase Enzyme (3 U L-1) 1, water X total reaction volume: 100, the two universal primers and 1.5 mM MgCl2. Amplification was done by initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 sec, annealing temperature of primers was 55°C for 30 sec and extension at 72°C for 2 min. Final extension was at 72°C for 15 min All PCR reagents were of Chromous Biotech, Ltd.
The PCR product were detected by using 1% agarose gel containing Ethidium bromide and the result recorded by UV transilluminator. Finally, the target PCR product was sliced and recovered. After purification, the PCR product was submitted for sequencing. Sequences have been deposited in GenBank.
Sequence analysis: Sequences were analyzed and aligned with other sequences deposited in Genbank by ClustalX (Thompson et al., 1997). The secondary structure and minimal free energy were calculated by Genebee tool (Mathews et al., 1999).
RESULTS AND DISCUSSION
Isolation and characterization bacterial of strains: The bacterial colonies differ greatly in their morphologies and these differences can help us in identifying different species of bacteria. The culture containing Streptococcus viridans, Staphylococcus aureus, Micrococcus sp., Moraxella sp., Citrobacter koseri, Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella sp., Propionibacterium sp., Staphylococcus epidermidis bacteria were and isolated from patients and identified by using biochemical tests. It shows that it was cocci shaped and Gram positive and Gram negative bacteria and it has glucose, lactose, sucrose fermentations (acid and gas), catalase enzyme activity (degradation of hydrogen peroxide) and non-acidic in nature.
Molecular characterization of bacteria: Molecular characterization of the bacterial strains was carried out by sequencing their 16S rDNA after its amplification and sequencing. Most strains showed high sequence identities with the same species on GenBank and some strains showed high identities with other species. This analysis made the identification of reference strains more reliable. The bacterial gene was sequenced and it deposited in the GenBank (NCBI) with the accession numbers JN378393, JN378392, HM204502, JQ039348, JN652127, JN652129, JQ039350, JN652128, JQ039349, HQ404365 for Streptococcus viridan, Staphylococcus aureus, Micrococcus sp., Moraxella sp., Citrobacter koseri, Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella sp., Propionibacterium sp., Staphylococcus epidermidis, respectively are shown in Table 1.
Sequence analyses and phylogenetic tree constructing: The 16S rRNA gene sequences of the bacterial strains studied by us were subject to multiple sequence alignment using ClustalX and the phylogenetic tree was drawn using Neighbour Joining plot. Multiple sequence alignment (Fig. 1) confirmed sequence conservation of the highly conserved regions of 16S rDNA of the causative bacteria of keratitis. The phylogenetic analysis of these genes from the phylogenetic tree (Fig. 2) shows that these genes are closely related.
| Table 1: | Bacteria isolated from keratitis patients |
 | |
 | |
 | |
 | |
 | |
 | |
 | |
| Fig. 1: | Multiple sequence alignment of 16S rDNA sequences of bacteria causing kertatitis |
GeneBee-secondary structure prediction: In the multiple alignment the algorithm used by GeneBee produces structures through the minimization of the potential energy of the system. The algorithm divides a sequence S, into subsequences (Sij) and calculates the minimal energy for two cases. W(i, j) defines the minimal free energy of all allowed structures formed from Sij and V(i, j) the minimum energy if Si and Sj pair with each other.
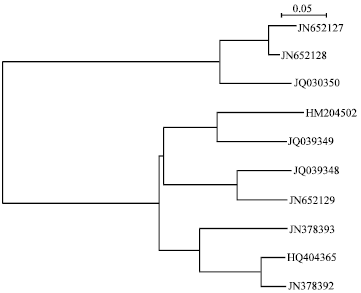 | |
| Fig. 2: | Phylogenetic analysis of 16S rDNA sequences of bacteria causing keratitis |
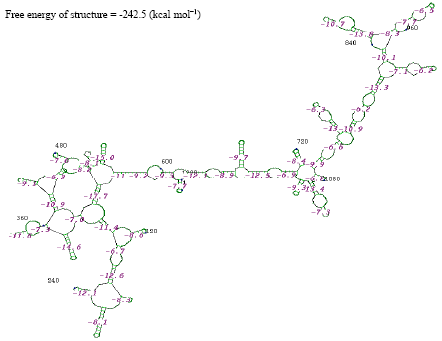 | |
| Fig. 3: | Structure of Streptococcus viridan |
The second term is set to infinity if Si and Sj cannot base pair. Starting with sequences of five nucleotides, W(i, j) and V(i, j) are calculated recursively for longer and longer sequences, selecting the optimal structure at each step. The final computation considering the entire sequence is the result. W(i, j) and V(i, j) are calculated based upon the stability of base pairs formed and the resulting structures: hairpin loops, stacking regions, bulge loops, interior loops and bifurcation loops as defined by Zuker and Stiegler (1981).
The thermostabilities of the secondary structure were calculated with online software (http://www.genebee.msu.su/services/rna2_reduced.html) (Fig. 3-12). The thermostability energy was calculated as -242.5, -74.4, -307.3, -275.2, -326.3, 46.7, -161.2, -234.8, -131.7 and -293.4 kcal mol-1, for Streptococcus viridians, Staphylococcus aureus, Micrococcus sp., Moraxella sp., Citrobacter koseri, Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella sp., Propionibacterium sp., Staphylococcus epidermidis, respectively (Table 2).
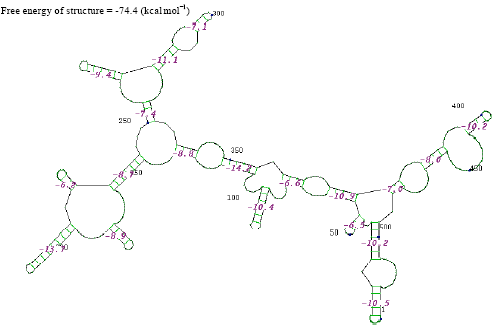 | |
| Fig. 4: | Structure of Staphylococcus aureus |
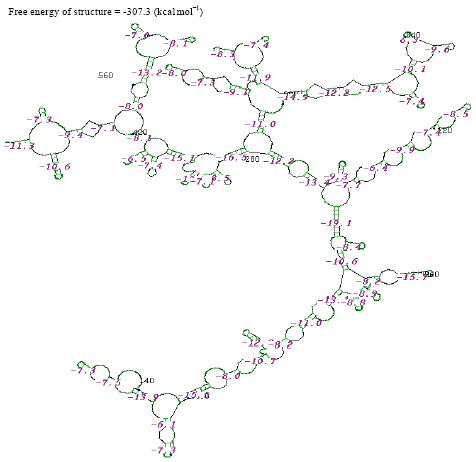 | |
| Fig. 5: | Structure of Micrococcus sp. |
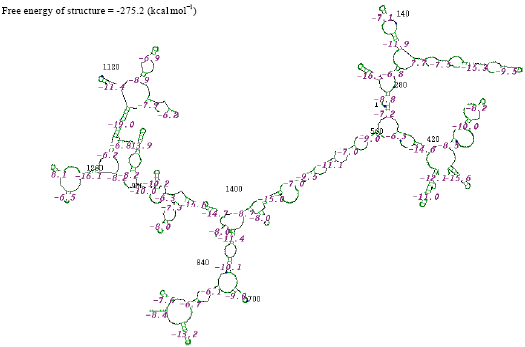 | |
| Fig. 6: | Structure of Moraxella sp. |
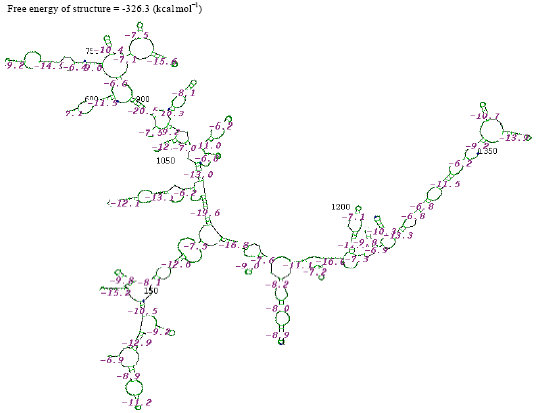 | |
| Fig. 7: | Structure of Citrobacter koseri |
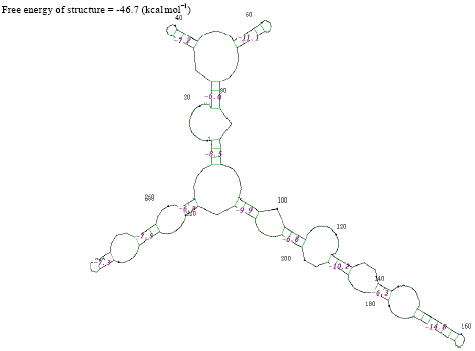 | |
| Fig. 8: | Structure of Acinetobacter baumannii |
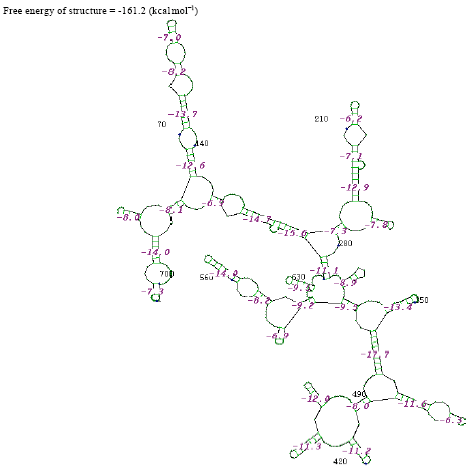 | |
| Fig. 9: | Structure of Pseudomonas aeruginosa |
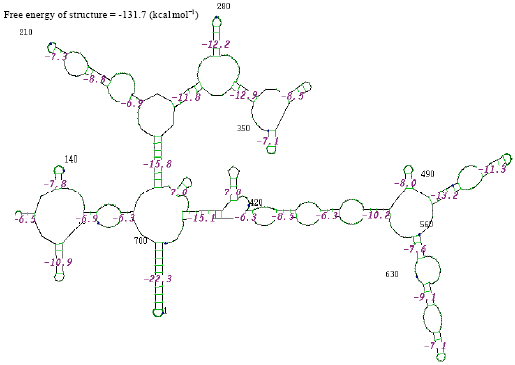 | |
| Fig. 10: | Structure of Propionibacterium sp. |
 | |
| Fig. 11: | Structure of Klebsiella sp. |
 | |
| Fig. 12: | Structure of Staphylococcus epidermidis |
| Table 2: | Secondary structure prediction of rDNA for bacterial sequences by using GeneBee tool |
 | |
The determination of the structure of the RNA molecule is very important in the determination of its activity.
In the secondary structure prediction of RNA the fold with the more negative free energy, is more stable. From the result, the Citrobacter koseri, (-326.3 kkal mol-1) has lower minimal free energy. When compared with others species whereas the Acinetobacter baumannii, (-46.7 kkal mol-1) has high minimal free energy when compared with other species. RNA molecule must be energetically stable to be able to maintain their structure and perform their functions.
rDNA-based analysis is a central method in microbiology, used as a method for bacterial strain identification. Molecular methods, including PCR, genotyping and automated DNA sequencing, are becoming more available and hence their adoption into routine medical microbiology is increasing. Therefore, 16S rDNA is known to be highly conserved in species and it could also be used for the verification of the thermodynamic stability on the basis of conserved secondary structure of RNA. The structure of RNA plays an important role in the life cycle of bacteria and provides the ability to understand evolution and stability (Zuker and Stiegler, 1981). Earlier studies reported that Molecular Characterization and Sequencing of a Gene Encoding Mannose Binding Protein in an Iranian Isolate of Acanthamoeba castellanii as a major agent of Acanthamoeba keratitis by PCR techniques (Niyyati et al., 2008).
In this study, we have characterized the bacteria causing keratitis and analyzed the phylogenetic relationship between them. The Multiple sequence analysis reveals that this sequence is highly conserved as all the organism share a region which is highly similar. The rDNA has also been modeled and their free energies have been predicted.
CONCLUSION
All bacterial species possess at least one copy of the 16S rDNA gene which contains highly conserved as well as hyper variable nucleic acid sequences. Therefore, the PCR targeting of the conserved nucleic acid sequence of 16S rDNA gene of bacterial isolate is used as a molecular tool to identify bacteria causing keratitis.
In conclusion, identification and sequencing of this important gene is the first step to pursue future research such as developing better therapeutic agents, immunization of population at risk or even developing a rapid diagnostic tool by PCR techniques. The sequence analysis of the gene fragment characterized in this study is still under investigation. This technique provides the opportunity to identify non-cultivable, uncommon, or even unknown causative bacteria.
REFERENCES
- Dunlop, A.A., E.D. Wright, S.A. Howlader, I. Nazrul, R. Husain, K. McClellan and F.A. Billson, 1994. Suppurative corneal ulceration in Bangladesh. A study of 142 cases examining the microbiological diagnosis, clinical and epidemiological features of bacterial and fungal keratitis. Aust. N.Z. J. Ophthalmol., 22: 105-110.
PubMed - Upadhyay, M.P., P.C. Karmacharya, S. Koirala, N.R. Tuladhar, L.E. Bryan, G. Smolin and J.P. Whitcher, 1991. Epidemiologic characteristics, predisposing factors and etiologic diagnosis of corneal ulceration in Nepal. Am. J. Ophthalmol., 111: 92-99.
PubMed - Asbell, P. and S. Stenson, 1982. Ulcerative keratitis. Survey of 30 year's laboratory experience. Arch. Ophthalmol., 100: 77-80.
PubMed - Liesegang, T.J. and R.K. Forster, 1980. Spectrum of microbial keratitis in South Florida. Am. J. Ophthalmol., 90: 38-47.
PubMed - Morlet, N. and M. Daniell, 2003. View 2: Empirical fluoroquinolone therapy is sufficient initial treatment. Br. J. Ophthalmol., 87: 1169-1172.
CrossRef - Jones, D.B., 1979. Initial therapy of suspected microbial corneal ulcers. II. Specific antibiotic therapy based on corneal smears. Surv. Ophthalmol., 24: 105-116.
PubMed - Hyndiuk, R.A., 1981. Experimental Pseudomonas keratitis. Trans. Am. Ophthalmol. Soc., 79: 541-624.
Direct Link - Bharathi, M.J., R. Ramakrishnan, S. Vasu, Meenakshi and R. Palaniappan, 2002. Aetiological diagnosis of microbial keratitis in South India - a study of 1618 cases. Indian J. Med. Microbiol., 20: 19-24.
PubMed - Thompson, J.D., T.J. Gibson, F. Plewniak, F. Jeanmougin and D.G. Higgins, 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25: 4876-4882.
CrossRefPubMedDirect Link - Mathews, D.H., J. Sabina, M. Zuker and D.H. Turner, 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288: 911-940.
CrossRefDirect Link - Zuker, M. and P. Stiegler, 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res., 9: 133-148.
CrossRefPubMedDirect Link - Niyyati, M., S. Rezaie, F. Rahimi, M. Mohebali, A.H. Maghsood, S.H. Farnia and M. Rezaeian, 2008. Molecular characterization and sequencing of a gene encoding mannose binding protein in an iranian isolate of Acanthamoeba castellanii as a major agent of Acanthamoeba keratitis. Iran. J. Publ. Health, 37: 9-14.
Direct Link