
Chandra Shekhar
Department of Biotechnology, Institute of Biomedical Education and Research, Mangalayatan University, Aligarh-202145, U.P., India
Kapil Dev
Department of Biotechnology, Institute of Biomedical Education and Research, Mangalayatan University, Aligarh-202145, U.P., India
Sitansu Kumar Verma
Department of Biotechnology, Institute of Biomedical Education and Research, Mangalayatan University, Aligarh-202145, U.P., India
Ajay Kumar
Department of Biotechnology, Institute of Biomedical Education and Research, Mangalayatan University, Aligarh-202145, U.P., India
Trends in Bioinformatics
Year: 2012 | Volume: 5 | Issue: 1 | Page No.: 14-24







ABSTRACT
Crimean-Congo Hemorrhagic Fever (CCHF) is a zoonotic viral disease that is asymptomatic in infected livestock but a serious threat to humans. This study is aimed at conducting the modeling of putative peptides which are suggested for vaccine development that is meant for evaluating epidemiological, clinical and laboratory characteristics of the patients diagnosed with Crimean-Congo hemorrhagic fever. In the present study, more reliable prediction of Major Histocompatibility Complex (MHC) peptide binding is based on the accurate determination of T-cell epitopes and hence the successful design of peptide and protein based vaccines. The importance of existing computational tools was used for prediction of peptide binding to Major Histocompatibility Complex (MHC) Class-I and Major Histocompatibility Complex (MHC) Class-II. With the availability of large sequence databases and computer aided design of peptide based vaccine, screening among billions of possible immune active peptides to find those likely to provoke an immune response was done. These peptides were selected by using different algorithms as Artificial Neuronal Network (ANN) and Support Vector Machine (SVM) for the T-cell epitope prediction and further characterized on the basis of binding affinity of peptide to HLA-alleles which can be finally used for the potential vaccine candidate development. A vaccine with specificity for a target population i.e., peptide based vaccine, in which small peptides derived from target proteins are used to provoke an immune reaction. Two nonameric epitopes (LRFGMLAGL) and (LLGIKCSFV) which exhibit good binding with MHC molecules and low energy minimization values providing stability to the peptide-MHC complex are reported here. These predicted peptides don’t have similarity with human proteome. These peptide could be used in designing a chimeric/subunit vaccine, however, these will further be tested by wet lab studies for a targeted vaccine design against Crimean-Congo hemorrhagic fever.
PDF Abstract XML References Citation
Received: May 27, 2011;
Accepted: July 06, 2011;
Published: May 21, 2012
How to cite this article
Chandra Shekhar, Kapil Dev, Sitansu Kumar Verma and Ajay Kumar, 2012. In-silico: Screening and Modeling of CTL Binding Epitopes of Crimean Congo Hemorrhagic Fever Virus. Trends in Bioinformatics, 5: 14-24.
DOI: 10.3923/tb.2012.14.24
URL: https://scialert.net/abstract/?doi=tb.2012.14.24
DOI: 10.3923/tb.2012.14.24
URL: https://scialert.net/abstract/?doi=tb.2012.14.24
INTRODUCTION
Crimean-Congo Hemorrhagic Fever (CCHF) virus is the member of family Bunyaviridae (genus Nairovirus). In 1940s this virus was first described when many cases of severe hemorrhagic fever arose among agricultural workers in the Crimean peninsula but this virus was firstly reported in the former Soviet Union in 1944 (Jain et al., 2011). After some years, a virus with similar pathogenesis was isolated in 1956 from patient in Congo, Africa and the virus was subsequently named as Crimean-Congo Hemorrhagic Fever (CCHF) virus (Jain et al., 2011). Crimean-Congo Hemorrhagic Fever (CCHF) has the most extensive geographic range of the medically significant tick-borne viruses, occurring in parts of sub-Saharan Africa, Asia, eastern Europe and the Middle East (Whitehouse, 2004). The countries widely affected in these areas include: Arabian Peninsula, Iraq, Pakistan and Xinjiang Province in northwest China (Ergonul, 2006). CCHF is a severe hemorrhagic fever in humans with a high fatality rate up to 30% (Ergonul, 2006; Vatansever et al., 2007). During the 21st century, recent outbreaks of CCHF virus were also reported in Gujarat, India.
The geographic distribution of CCHF virus cases corresponds most closely with the distribution of Hyalomma ticks, hosted on the migratory birds suggesting their principle vector role (Vatansever et al., 2007; Whitehouse, 2004). Some others species of Dermacentor and Rhipicephalus genera have also been shown to be capable of transovarial transmission. CCHFV is a member of large family of negative stranded RNA viruses denoted by Bunyaviridae. The family consists of more than 300 viral species and is subdivided into five genera: Orthobunyavirus, Hantavirus, Phlebovirus, Tospovirus and finally Nairovirus (Nichol et al., 2005). CCHFV has three segments of negative sense RNA viz. S, M and L which minimally encode the virus nucleocapsid, glycoproteins and polymerase proteins, respectively. M-RNA segment of CCHFV plays a major role in the immune response. In addition, members of the genera Bunyavirus, Phlebovirus and Tospovirus also encode a nonstructural glycoprotein referred to as NSM (Honig et al., 2004; Kinsella et al., 2004; Meissner et al., 2006). But, the complete information about the M RNA fragment is not available. The M gene is responsible for immunity and pathogenicity as well as for vaccine development. The nucleotide sequences of the M RNA genome segment of CCHFV strains isolated from Xinjiang province was determined to define the molecular variability among CCHFV strains, in China. Examination of their expected amino acid sequences with the respective sequences of the orientated protein was also carried out (Meissner et al., 2006). Epitope based vaccine provide a new strategy for the prophylactic and therapeutic application of pathogen specific immunity (Zinkernagel and Hengartner, 2004). This strategy requires the identification and selection of promiscuous T-cell epitopes important for cytolytic and regulatory response to pathogens that helps to the vaccine development (Esser et al., 2003; Brusic and Agust, 2004; Pulendran and Ahmed, 2006).
The progression of Congo Hemorrhagic Fever is very rapid with clinical features as flu like symptoms appears during primary 3 days of infection. After one week symptoms get resolved, 75% cases which have sign of hemorrhage, thrombosis of vessels to extremities and leading to death within 5 to 7 days. No procurement found for this viral infection. Researches for the development of effective vaccine against CCHF virus require understanding of immune response. Viral immune response is associated with MHC protein and T-lymphocytes. MHC is of two types: MHC Class I and MHC Class II (Rammensee et al., 1999). MHC initially recognizes the viral antigenic epitopes present on T-cells for neutralization. MHC Class I present the antigenic epitopes to CD8+ T-cells and MHC Class II present to CD4+ T-cells for viral antigen degradation (Adams and Koziol, 1995; Berman et al., 2000). CD8 T-cells also known as cytotoxic T-cells (CTL), maximum viral infections by initially recognizing and their subsequent killing infected cells and secreting cytokines. CD4 T-cells known as helper cells that play very important role in growth factor releasing and signaling for generation and maintenance of CD8 T-cells (Zielkiewicz, 2005).
T-cells recognize the antigens only when they are associated with MHC surface glycoproteins exposed on surface of all vertebrate cells. In this communication, online bioinformatics tools were used and the targeted protein of CCHFV was analyzed, to identify the putative T-cell epitopes for the formation of peptide based vaccine. The vaccine of Congo virus is not yet available. The CCHF vaccine development is very difficult because it requires the known pathogenic human host and it is also difficult to grow the virus in culture medium. The significance of this modern approach is, it reduces the time and risk of pathogenesis; in order to overcome the problems of attenuated vaccine development. Epitope based vaccine consists of short peptide sequence which is derived from small part of virulence protein. Antigenic determinants are present in certain part of the vaccine peptide sequence. This vaccine should cover the Human Leukocyte Antigen (HLA) haplotypes of the target population, be effective against a wider spectrum of Congo virus strains and not have any self-effective epitopes and produce effective immune response (Eswar et al., 2007). A No. of computational tools are now available for prediction of T-cell epitopes (Parida et al., 2007; Shakyawar et al., 2011), all overlapping nonamers of CCHFV to Human HLA Class I molecules have been analysed. The selected peptides have been modeled on corresponding HLA to validate the binding prediction. Some peptide identified from Bioinformatics and Molecular Analysis Section (BIMAS) and SYFPEITHI that binds to Class I HLA are also revealed. A No. of peptides were chosen for structural modeling on the bases of their binding affinity to Class I alleles by multiple analytical tools, since, the putative peptide is predicted to form stable complex with HLA allele through molecular modeling and it have not any identical peptide of Human proteome cross checked by HLApred (Berman et al., 2000). The aim of the study is to suggest these peptides as potential vaccine candidate development.
MATERIAL AND METHODS
The in silico study was conducted at Department of Biotechnology, Mangalayatan University, Aligarh from Nov, 2010 to May, 2011.
Virus and protein: RNA-dependent RNA polymerase (ACM78472.1), nucleocapsid protein (ABB30042.1) and nucleoprotein (BAE80107.1) are the protein of Crimean-Congo Hemorrhagic Fever Virus (CCHFV) strain isolated from Xinjiang province available in the NCBI Protein data base and hence it is used for this analysis.
Physical properties of the selected proteins: Bioinformatics tools were used for the analysis of proteome of CCHF virus. The protein sequences of were retrieved from www.ncbi.nlm.nih.gov. The expected molecular weight, highly repeated amino acids (%) of repetition, least repeated amino acid and isoelectric point (pI) values were calculated using ExPaSy (http://www.expasy.org/) (Kyte and Doolittle, 1982; Shehzadi et al., 2011).
MHC-Class I binding epitope prediction: All of these targeted proteins of the CCHF virus strain found in Xinjiang namely RNA-dependent RNA polymerase, nucleocapsid protein and nucleoprotein were analyzed for the Cytotoxic T-lymphocytes (CTL) epitopes using several algorithms. BIMAS online tool was used to analyze binding of all consensus peptides with 33 human HLA allele that which helps to identify those peptides in the targeted proteins with high affinity promiscuous epitopes that binds to HLA (Parker et al., 1994). The binding affinity (T1/2) value is based on the half time (min) of dissociation of β2 microglobulin from HLA. The T1/2 value was set at cutoff T1/2 ≥100 for peptide selection, other several algorithms based tools viz. Propred1, SYFPEITHI and Propred are also used for prediction of putative T-cell epitopes (Singh and Raghava, 2001).
Propred1 is matrix-based method that allows prediction of MHC binders for various alleles based on the multiplication and additional matrices, proteosome cleavage site, simultaneously. This is based on the observations made in previous studies which demonstrate that MHC binders having proteosome cleavage site at their C terminus have high potency to become T-cell epitopes. (Singh and Raghava, 2001). Endogenously synthesized peptides of 9-11 amino acids of HLA class I molecules get interacted with T-cell receptor of T-cells on the surface of infected cells. The presence of allele-specific amino acid motifs has been demonstrated by sequencing of peptides eluted from MHC molecules (Lund et al., 2002).
Structure-based modeling of T-cell epitope: Molecular modeling and structural analysis (Chaitra et al., 2005) were performed for the detection of binding peptides to their respective class I HLA alleles. Sample peptides of high affinity binders for a few alleles where structures are known (A 0201, A2, A 2402, B 1501, B 2705, B 2709, B 3501, B 4403, B 4405, B 5101, B 5301) were modeled employing their respective structural templates (1AO7, 1AKJ, 2BCK, 1XR8, 1HSA, 1UXW, 1A1M, 1SYS, 1SYV, 1A1O, 1EFX, 1IM9 and 1QQD). Two peptides viz. LLGIKCSFV and LRFGMLAGL were selected with the help of scoring based algorithms of BIMAS (Parker et al., 1994; Parida et al., 2007). These peptides have higher binding affinity. These selected peptides with highest and lowest T(1/2) were modeled on to their respective structural templates and the complexes were subjected to energy minimizations (Vani et al., 2006). The binding of the peptides was estimated by analyzing the intra-molecular hydrogen bonds, electrostatic, van der Waals and hydrophobic interactions with the protein residues in the vicinity.
The Modeller (Eswar et al., 2007) was used for the designing of the structures of those alleles whose structures were not available in the PDB server while the CPH model server (Nielsen et al., 2010) was used to design the structures of the predicted binding peptides. After designing the structures docking of selected alleles and peptides was performed with the help of Autodock. This was done to out the energy minimization (Morris et al., 1998; Namasivayam and Gunther, 2007; Amir et al., 2010) and then PMV (Python Molecular Viewer) was used for the visualization of Binding, position, H-bonding between the selected peptides and alleles.
RESULT
In this present study, three putative proteins of CCHF virus were used for the physicochemical analysis (Stevenson et al., 2007) such as molecular weight, isoelectric point (pI value) and antigenic nature. The RNA-Dependent RNA Polymerase protein has the highest molecular weight of about 447836.7 KDa which consists of Leucine (L) a neutral nonpolar amino acid residue has the highest percentage of repetition (12.4%). The least repeated residue of L-segmented protein of CCHFV is a nonpolar Tryptophan (W) (0.9%). The M-segment viral peptide encoded by nucleocapsid protein is NCM (53955.5 KDa molecular weight) comprising 482 amino acid residues. Lysine (K) has highest percentage and Cysteine (C) is the least repetitive amino acid residues(1.2%) of this protein; another targeted protein has the lowest molecular weight of about 8223.4 KDa (i.e., S-segment encoded the nucleoprotein), Lysine(K) has the highest percentage of repetition (13.7%), Histidine (H) and Proline (P) are the least repeated amino acid residues (1.4%) of nucleoprotein. The physicochemical properties of putative proteins were given in Table 1. The pI value of any protein indicates the stability of protein in that particular isoelectric point. Isoelectric points of these proteins were ranged between 7.14 to 9.48.
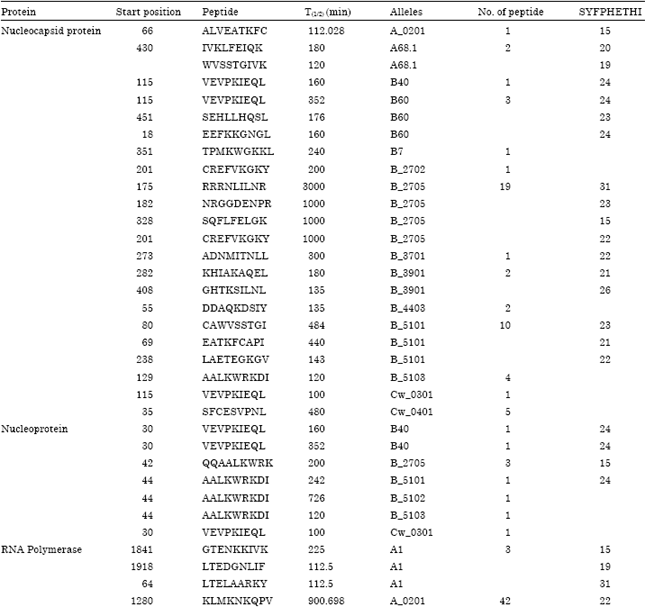
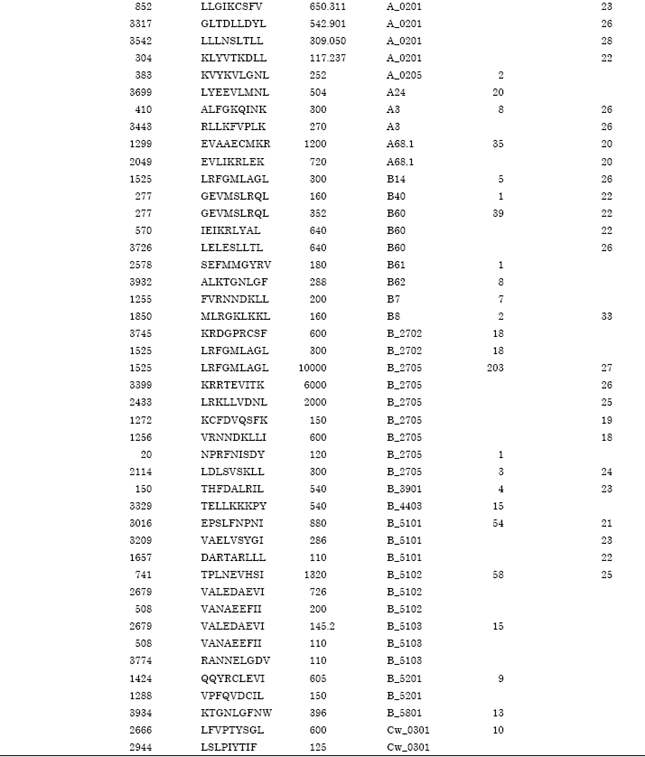
Binding specificity of promiscuous T-cell epitopes to HLA class-I molecules: The prediction of epitopes with their position and corresponding promiscuous HLA alleles by using different tools has been summarized in Table 2.
| Table 1: | It comprises the data of CCHFV proteins, molecular weight and percentage of highly repeated and least repeated amino acid residues in individual protein. The percentage of amino acid residues gives an outlook for their pI value and their probability of incidence in the antigenic epitopes |
 | |
| L*(Lucine) and W# (Tryptophan) are non-polar anchor residue for HLA predicted epitopes. K**(Lysine) is an anchor residue and also for HLA predicted epitopes. H/P***(Histidine/Proline) are least repeated amino acids | |
| Table 2: | The predicted peptides from target protein binds to different HLA class I alleles (BIMAS T(1/2)≥100 and SYFPEITHI value≥15) |
  | |
The promiscuity of binding of a peptide to HLA alleles is important since inclusion of such peptides in the vaccine construct provides a greater population coverage which helps to short out the promiscuous peptide that needs to be in vaccine developments (Herrera et al., 2010) .
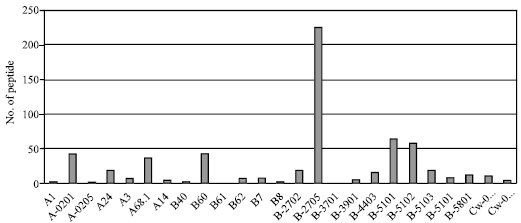 | |
| Fig. 1: | This conservation plot represents the number of peptides of all three protein of CCHFV that binds to 33 Class I HLA alleles at cutoff T(1/2) ≥100. B_2705, B_5101, B_5102, A_0201, B_60, A_68.1 are some strong binding alleles which bind to most of the peptides and also shown by the tallest bar. HLA alleles A3, A24, B14, B62, B7, B_2702, B_4403, B_5103, B_5201, B_5801 and Cw_0301, Cw_0401 bind to the less No. of predicted epitopes |
BIMAS is the immunoinformatics tool freely available to be used for prediction of the antigenic epitopes in the complete protein with more effective and accurate prediction of MHC binding affinity (i.e., T (1/2) value). The binding analysis of all conserved nonamers of all three consensus CCHFV proteins to 33 HLA class I alleles at different binding affinities.
Total 71 epitopes were predicted against 33 alleles of MHC Class I by using the tool BIMAS. The maximum number of epitopes were represented by RNA dependent-RNA polymerase protein comprising 65% of all MHC Class I predicted epitopes, 29.5% of MHC Class I predicted epitopes from nucleocapsid protein and minimum number of epitopes from nucleoprotein 5.6% (Table 2). LLGIKCSFV, LRFGMLAGL, EPSLFNPNI and SQFLFELGK are the promiscuous binders of MHC Class I alleles. In case of nucleocapsid protein RRRNLLLNR and CAWVSSTGI are the best binders in terms of quantitative scores of HLA alleles (MHC Class I) coverage. For the nucleoprotein not also have the epitope of good quantity covering HLA alleles available in BIMAS. Out of these 33 HLA alleles B_2705 is capable for binding to the highest number of predicted promiscuous epitopes all proteins show the tallest bar in Fig. 1. Other HLA alleles binding to the less number of promiscuous epitopes are also shown in same figure.
Conserved epitope of CCHF virus protein: It is important to identify those peptides which are conserved across the various strains of CCHF virus and in this study that has been shown for the conserved peptides present in the constituent proteins of CCHF virus.
The analysis reveals that there are number of suitable peptides from RNA dependent-RNA polymerase which may be included in the construction of poly epitopes T-cell vaccine (Parida et al., 2007). Some of the conserved peptides (LLGIKCSFV, EPSLFNPNI, LRFGMLAGL and SQFLFELGK) with class I presentation potential along with their interaction energies are, -29.31, -26.85, -35.05 and -23.97 (in kcal mol-1), respectively given in Table 3. High T(1/2) and interaction energy indicate high HLA binding affinity. These peptides are promiscuous HLA binders. It will be useful to include these peptides in a chimeric constructs containing both cytotoxic and helper epitopes. It is expected that though this T-cell vaccine would not prevent CCHF virus infection, it would aid in quick clearance of the virus and prevent the severe infection (Parida et al., 2007).
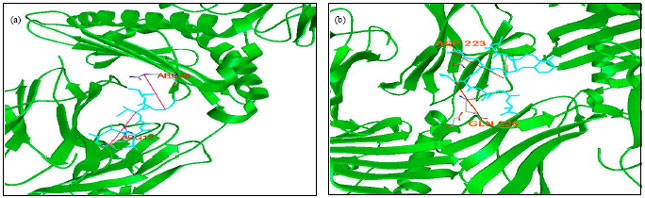 | |
| Fig. 2: | The peptide binding to the HLA class I molecule. The peptides (shown in green colour) predicted to a have very high affinity for the allele A_0201 and B_2705 modeled on to the crystal structure (1AO7 and 1 HAS) based on the position of the peptide. Potential hydrogen bonds are shown. The high binder peptide (a) LLGIKCSFV and (b) LRFGMLAGL are derived from the CCHF Virus |
| Table 3: | The conformational properties of the peptides with efficient binding energy and present on the variable regions of predicted peptide as investigated by molecular dynamics simulation using Autodock tool v3.0 |
 | |
DISCUSSION
It is reported that, the primary function of T-Cell vaccine is to generate CTLs to degrade the virus infected cells. The viral antigens released when get lysed are capable of stimulating antibody response against these antigens get leading to neutralization of reinfecting and residual viruses in the system. Since these events take place in during the incubation phase of the virus infection, if any. It is likely to be very mild. Beside the idiotype and anti-idiotypic antibody cascades generated by T-Cell epitopes would reinforce T-Cell memory (Lal et al., 2006; Nayak et al., 2001, 2005; Mohabatkar and Mohammadzadegan, 2007).
The characterization of putative peptides on the basis of antigenic variability depends on the surface exposed regions of target CCHFV protein revealed that 6 of the total 71 predicted epitopes were present. Out of six short listed peptides, four peptides were chosen here for their further characterization on the basis of their energy minimization value. Moreover with the SYFPEITHI, it scored high with a value of 27 and 23 for the binding to HLA alleles like B_2705 and A_0201 corresponding to their short listed peptides LRFGMLAGL and LLGIKCSFV, respectively. Figure 2a and b illustrates the interaction of these two peptides with their respective alleles (Kavita et al., 2010). The resulting peptides LRFGMLAGL, of RNA polymerase protein binds to HLA B_2705. It is seen to make two hydrogen bonds from its arginine in the 2nd position to asparagine at position 223 of the B_2705 and leucine in the 1st position to a glutamine at position 226 of this allele. Similarly the peptide LLGIKCSFV, of same protein binds to HLA A_0201. It also makes two hydrogen bonds from its lysine in the 5th position to a arginine at position 48 of the allele A_0201 and glycine in the 3rd position to a arginine at position 12th of this allele with its high affinity (Parida et al., 2007; Tambunan and Parikesit, 2010).
It must be noted that MHC class I a peptides have preference for hydrophobic or positively charged amino acid residues at carboxyl end for proper biding in pockets (Brusic et al., 2002). The screening in this work also listed two more peptides TPLNEVHSI and GEVMSLRQL that were presented on class I allele of RNA dependent-RNA polymerase and scored 1320 and 352 with BIMAS. These epitopes were, however, not included for simulation analysis. Simulation studies of the epitope LRFGMLAGL and LLGIKCSFV formed stable MHC-peptide complexes with the energy minimization of -35.05 and -29.31 (kcal mol-1), respectively. The other two peptides EPSLFNPNI and SQFLFELGK identified in the present study were found to be antigenically variable with energy minimization value of -26.85 and -23.97 (kcal mol-1), respectively (Kavita et al., 2010). This can possibly be targeted for designing of vaccine against CCHF virus strains of Xinjiang province.
CONCLUSION
The screening of putative epitopes using bioinformatics tools thus suggests that protein RNA dependent-RNA polymerase of CCHFV could be used for preparation of immunological constructs. Molecular simulation and binding tests also suggest that the two nonameric epitopes LRFGMLAGL and LLGIKCSFV predicted and reported for the first time have considerable binding with MHC molecules and low energy minimization values providing stability to the peptide-MHC complex. These peptide construct will further undergo wet lab studies, for the development of targeted vaccine against CCHF virus strains. Using a similar approach the short listed candidate epitopes for vaccine design using other proteins can also be targeted that would reduce time and experimental expense.
ACKNOWLEDGMENTS
The authors are grateful to Prof. Ashok Kumar (Dean, I.B.M.E.R, Mangalayatan University Aligarh, U.P., India) for providing necessary facilities and encouragement. The authors are also thankful to all faculty members of the Institute of Biomedical Education and Research, Mangalayatan University Aligarh, U.P., India for their generous help and suggestions during the course of experimental work and manuscript preparation.
REFERENCES
- Adams, H.P. and J.A. Koziol, 1995. Prediction of binding to MHC class I molecules. J. Immunol. Methods, 185: 181-190.
PubMed - Berman, H.M., J. Westbrook, Z. Feng, G. Gilliland and T.N. Bhat et al., 2000. The protein data bank. Nucl. Acids Res., 28: 235-242.
CrossRefDirect Link - Brusic, V. and J.T. August, 2004. The changing field of vaccine development in the genomics era. Pharamacogenomics, 5: 597-600.
PubMed - Brusic, V., N. Petrovsky, G. Zhang and V.B. Bajic, 2002. Prediction of promiscuous peptides that bind HLA class I molecules. Immunol. Cell Biol., 80: 280-285.
PubMed - Chaitra, M.G., S. Hariharaputran, N.R. Chandra, M.S. Shaila and R. Nayak, 2005. Defining putative T cell epitopes from PE and PPE families of proteins of mycobacterium tuberculosis with vaccine potential. Vaccine, 23: 1265-1272.
Direct Link - Esser, M.T., R.D. Marchese, L.S. Kierstead, L.G. Tussey, F. Wang, N. Chirmule and M.W. Washabaugh, 2003. Memory T cells and vaccines. Vaccine, 21: 419-430.
CrossRef - Honig, J.E., J.C. Osborne and S.T. Nichol, 2004. Crimean-Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology, 321: 29-35.
CrossRef - Jain, H., S.K. Gediya, D.K. Thakkar, N.Y. Mansuri, K.M. Ashar and T.Y. Pasha, 2011. Crimean congo hemorrhagic fever virus. JPBMS, 5: 1-6.
Direct Link - Kavita, S., C. Priyanka and M. Namrata, 2010. Bioinformatics approach for screening and modeling of putative T cell epitopes from Por B protein of Neisseria meningitides as vaccine constructs. Indian J. Biotechnol., 9: 351-359.
Direct Link - Kinsella, E., S.G. Martin, A. Grolla, M. Czub, H. Feldmann and R. Flick, 2004. Sequence determination of the Crimean-Congo hemorrhagic fever virus L segment. Virology, 321: 23-28.
CrossRef - Kyte, J. and R.F. Doolittle, 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol., 157: 105-132.
PubMed - Lal, G., M.S. Shaila and R. Nayak, 2006. Activated mouse T cells downregulate, process and present their surface TCR to cognate anti-idiotypic CD4+ T cells. Immunol. Cell Biol., 84: 145-153.
PubMed - Meissner, J.D., S.S. Seregin, S.V. Seregin, O.I. Vyshemirskii and E.I. Samokhvalov et al., 2006. A variable region in the Crimean-Congo hemorrhagic fever virus L segment distinguishes between strains isolated from different geographic regions. J. Med. Virol., 78: 223-228.
CrossRef - Morris, G.M., D.S. Goodsell, R.S. Halliday, R. Huey, W.E. Hart, R.K. Belew and A.J. Olson, 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem., 19: 1639-1662.
CrossRefDirect Link - Namasivayam, V. and R. Gunther, 2007. pso@autodock: A fast flexible molecular docking program based on Swarm intelligence. Chem. Biol. Drug Des., 70: 475-484.
PubMed - Eswar, N., B. Webb, M.A. Marti-Renom, M.S. Madhusudhan and D. Eramian et al., 2007. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein. Sci.
CrossRef - Nayak, R., G. Lal and M.S. Shaila, 2005. Perpetuation of immunological memory: Role of serum antibodies and accessory cells. Microbes Infect., 7: 1276-1283.
CrossRef - Nayak, R., S. Mitra-Kaushik and M.S. Shaila, 2001. Perpetuation of immunological memory: A relay hypothesis. Immunology, 102: 387-395.
CrossRef - Nichol, S.T., B.J. Beaty, R.M. Elliott, R.W. Goldbach, A. Plyusnin and R.B. Tesh, 2005. The Bunyaviridae. In: Virus Taxonomy: Classification and Nomenclature of Viruses: Eighth Report of the International Committee on Taxonomy of Viruses, Fauquet, C.M., M.A. Mayo, J. Maniloff, U. Desselberger and L.A. Ball (Eds.). Elsevier/Academic Press, London, United Kingdom, pp: 695-716.
- Nielsen, M., C. Lundegaard, O. Lund and T.N. Petersen, 2010. CPHmodels-3.0--remote homology modeling using structure-guided sequence profiles. Nucl. Acids Res., 38: W576-W581.
PubMed - Parida, R., M.S. Shaila, S. Mukherjee, N.R. Chandra and R. Nayak, 2007. Computational analysis of proteome of H5N1 avian influenza virus to define T cell epitopes with vaccine potential. Vaccine, 25: 7530-7539.
CrossRef - Parker, K.C., M.A. Bednarek and J.E. Coligan, 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol., 152: 163-175.
PubMed - Pulendran, B. and R. Ahmed, 2006. Translating innate immunity into immunological memory: Implications for vaccine development. Cell, 124: 849-863.
PubMed - Rammensee, H.G., J. Bachmann, N.P.N. Emmerich, O.A. Bachor and S. Stevanovic, 1999. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics, 50: 213-219.
CrossRefDirect Link - Shehzadi, A., S. Ur Rehman and M. Idrees, 2011. Promiscuous prediction and conservancy analysis of CTL binding epitopes of HCV 3a viral proteome from Punjab Pakistan: An in silico approach. Virol. J., 8: 55-55.
PubMed - Singh, H. and G.P.S. Raghava, 2001. ProPred: Prediction of HLA-DR binding sites. Bioinformatics, 17: 1236-1237.
CrossRefPubMedDirect Link - Tambunan, U.S.F. and A.A. Parikesit, 2010. In silico design of drugs and vaccines for dengue disease. Trends Bioinform., (In Press).
Direct Link - Vani, J., M.S. Shaila, N.R. Chandra and R. Nayak, 2006. A combined immuno-informatics and structure-based modeling approach for prediction of T cell epitopes of secretory proteins of Mycobacterium tuberculosis. Microbes Infect., 8: 738-746.
Direct Link - Whitehouse, C.A., 2004. Crimean-congo hemorrhagic fever. Antiviral Res., 64: 145-160.
CrossRefPubMedDirect Link - Zielkiewicz, J., 2005. Structural properties of water: Comparison of the SPC, SPCE, TIP4P and TIP5P models of water. Chem. Phys., 123: 1-6.
CrossRef - Zinkernagel, R.M. and H. Hengartner, 2004. On immunity against infections and vaccines: Credo 2004. Scand. J. Immunol., 60: 9-13.
PubMedDirect Link - Amir, A., M.A. Siddiqui, N. Kapoor, A. Arya and H. Kumar, 2011. In silico molecular docking of influenza virus (PB2) protein to check the drug efficacy. Trends Bioinform., 4: 47-55.
CrossRefDirect Link - Stevenson, D.G., F.J. Eller, J.L. Jane and G.E. Inglett, 2007. Starch structures and physicochemical properties of a novel β-glucan-enriched oat hydrocolloid product with and without supercritical carbon dioxide extraction. Am. J. Food Technol., 2: 248-256.
CrossRefDirect Link - Herrera, D.I.M., J.A.M. Morales, A.E.P. Cardena, B. Molina Sanchez and M.A.R. Chessani et al., 2010. Use of RB51 vaccine for small ruminants brucellosis prevention, in Veracruz, Mexico. Int. J. Dairy Sci., 5: 10-17.
CrossRefDirect Link - Mohabatkar, H. and R. Mohammadzadegan, 2007. Computational comparison of T-cell epitopes of gp120 of Iranian HIV-1 with different subtypes of the virus. Pak. J. Biol. Sci., 10: 4295-4298.
CrossRefPubMedDirect Link - Shakyawar, S.K., A. Goyal and V.K. Dubey, 2011. Database of In silico predicted potential drug target proteins in common bacterial human pathogens. Am. J. Drug Discovery Dev., 1: 70-74.
CrossRefDirect Link