
A. Amir
Department of Biotechnology, Meerut Institute of Engineering and Technology, N.H. 58, Delhi-Roorkee Highway, Baghpat Road Bypass Crossing,Meerut-250005, UP, India
M. A. Siddiqui
Department of Biotechnology, Meerut Institute of Engineering and Technology, N.H. 58, Delhi-Roorkee Highway, Baghpat Road Bypass Crossing,Meerut-250005, UP, India
N. Kapoor
Department of Biotechnology, Meerut Institute of Engineering and Technology, N.H. 58, Delhi-Roorkee Highway, Baghpat Road Bypass Crossing,Meerut-250005, UP, India
A. Arya
Department of Biotechnology, Meerut Institute of Engineering and Technology, N.H. 58, Delhi-Roorkee Highway, Baghpat Road Bypass Crossing,Meerut-250005, UP, India
H. Kumar
Department of Biotechnology, Meerut Institute of Engineering and Technology, N.H. 58, Delhi-Roorkee Highway, Baghpat Road Bypass Crossing,Meerut-250005, UP, India
Trends in Bioinformatics
Year: 2011 | Volume: 4 | Issue: 1 | Page No.: 47-55







ABSTRACT
The aim of this study was to analyze and model the 3D structure PB2 protein of influenza A viruses of subtype H5N1 and predicting the most effective drug against influenza virus (subtype H5N1) from a list of available drugs by targeting PB2 protein. This was done first by database search of PB2 protein sequence which results in a sequence of 759 amino acids (Acc. No. ACZ58135). 3D structure of PB2 protein was not available in PDB database, therefore template structure (PDB ID: 2vqzD) with 92.727% sequence identity was selected. Homology model was constructed using Swiss Model and validated using PROCHEK. Ramchandran plot analysis shows 87.1% of the residues in the most favored region. The model was finally docked with three different drugs namely Rimantadine, Amantadine and Zanamivir. From the docking result it was observed that the drug rimantadine had the least binding energy and considered as the most effective drug against PB2 protein. The study is unique in nature as there is no known report available about the homology modeling of PB2 protein as well as its docking with known drugs. All work was done using in silico approaches.
PDF Abstract XML References Citation
Received: April 17, 2010;
Accepted: May 31, 2010;
Published: June 29, 2011
How to cite this article
A. Amir, M. A. Siddiqui, N. Kapoor, A. Arya and H. Kumar, 2011. In silico Molecular Docking of Influenza Virus (PB2) Protein to Check the Drug Efficacy. Trends in Bioinformatics, 4: 47-55.
DOI: 10.3923/tb.2011.47.55
URL: https://scialert.net/abstract/?doi=tb.2011.47.55
DOI: 10.3923/tb.2011.47.55
URL: https://scialert.net/abstract/?doi=tb.2011.47.55
INTRODUCTION
Influenza viruses belong to the member of orthomyxo-viridae family, having a single-standard, negative sense, segmented RNA genome in an enveloped virion (Anwar et al., 2006; Ahn et al., 2006). The genome of influenza viruses have 8 RNA segments encoding 10 proteins including two surface glycoprotein, haemaggulutinin (HA), neuraminidase (NA), nucleoproteins (NP), three polymerase proteins (PA, PB1, PB2) two matrix (M1, M2) and nonstructural proteins (NS1, NS2). Influenza viruses are classified as types A, B and C based on the antigenic properties of nucleoprotein (NP) and matrix (M1) (Kamal et al., 2007; Suzuki and Nei, 2002). Avian influenza is caused by a type-A virus and it is further classified into subtype based on two surface glycoprotein, haemagglutinin (HA) and neuraminidase (NA). There are about 16 known HA and 9 known NA in type-A viruses.
The highly pathogenic avian influenza H5N1 virus, which is panzootic in poultry, continues to spread and pose a major challenge to animal and human health (Epstein et al., 2002; WHO, 2007). Since pandemic influenza virus has its origins in avian influenza viruses (Webster et al., 1992), H5N1 virus has to be considered a potentially serious pandemic threat. New influenza virus pandemics in the 21st century are a certainty, but whether H5N1 will be the next pandemic virus is far from certain. What is already true, however, is that H5N1 viruses are taking a huge toll on the poultry industry in many developing countries and this directly or indirectly impacts both economic and social wellbeing. The potential impact of H5N1 virus (and human reaction to its spread) on wildlife and ecology has received less attention but is also worthy of consideration (Kamal et al., 2007).
H5N1 virus consist a PB2 protein which is a critical component of the viral polymerase. The polymerase protein PB2 is 759 residues long. The polymerase complex is formed protein subunit interactions with the PB1 subunit (Digard et al., 1989). PB2 interacts with NP and not with PA (Biswas et al., 1998; Zurcher et al., 1996). This subunit plays an important role in transcription of mRNA by binding with the 5’ methylated cap of pre-mRNA in host cells for providing primers to viral mRNA synthesis (Nakagawa et al., 1995). Two hypothetical domains (residues 242-280 and 538-577) identified in PB2 are hypothesized for viral binding to host cells (Honda et al., 1999).
IN SILICO TECHNIQUES TO RAPIDLY IDENTIFY THE DRUG EFFECTIVENESS
The cost of research and development in the pharmaceutical industry has been rising steeply and steadily to achieve any process in the last decade, but the amount of time required for bringing new products to market remains around ten to fifteen years. In silico technique is an inexpensive technique that shortens the length of time spent in checking the efficacy of drugs. Important factors in this context include homology modeling of virus protein (Marti-Renom et al., 2000), in silico molecular docking of virus protein PB2 with drugs (Kitchen et al., 2004). In the present study, we model H5N1 virus PB2 protein using Swiss model server followed by docking against known inhibitors using EXOME Horizon.
MATERIALS AND METHODS
This in silico study was conducted at Department of Biotechnology, Meerut Institute of Engineering and Technology, Meerut during Sep., 2009 to Jan, 2010. The methodology was as follows:
Retrival of data set: The PB2 protein sequence of influenza virus subtype H5N1 was retrieved from Genome directory INFLUENZA at NCBI (ftp://ftp.ncbi.nih.gov/genomes/INFLUENZA).
Homology modeling of PB2 protein: Homology modeling predicts the three dimensional structure of a given protein sequence (target) based on an alignment to one or more known protein structures (template). In general, 30% sequence identity is required to generate an useful model. SWISS-MODEL (http://swissmodel.expasy.org/) is a fully automated protein structure homology-modeling server, accessible via the ExPASy (www.expasy.ch) web server. The purpose of this server is to make Protein Modelling accessible to all biochemists and molecular biologists Worldwide. It provides several levels of user interaction through its World Wide Web interface. In automated mode only an amino acid sequence of protein is submitted to build a 3D model. Template selection, alignment and model building are done completely automated by the server. Homology modeling was performed to find best template of PB2 protein by using Swiss model (Gasteiger et al., 2005).
Model evaluation and validation: The predicted 3D model was evaluated and validated using Structure Analysis and Verification Server (SAVES) (http://nihserver.mbi.ucla.edu/SAVES/) using PROCHECK. SAVES is running by a research group at UCLA. It provides the facility to check the stereochemical quality of a protein structure by analyzing residue-by-residue geometry, determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) by assigned a structural class based on its location and environment (alpha, beta, loop, polar, nonpolar etc.) and comparing the results to good structures and overall structure geometry. The overall stereochemical quality of the protein was assessed by Ramachandran et al. (1963) plot analysis. Discovery Studio Visualizer (Accelrys) is a comprehensive software suite for analyzing was used to manipulate the models based on residue interactions, energy minimization and steric hinderance.
Drugs selection: The DrugBank database (http://www.drugbank.ca) is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e., chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e., sequence, structure and pathway) information. The database contains nearly 4800 drug entries including >1,350 FDA-approved small molecule drugs, 123 FDA-approved biotech (protein/peptide) drugs, 71 nutraceuticals and >3,243 experimental drugs. Additionally, more than 2,500 non-redundant protein (i.e., drug target) sequences are linked to these FDA approved drug entries. To check the drug effectiveness 3 drugs, rimantadine, amantadine and zanamivir has been searched using Drug Data Bank. The pdb file of each drug has been downloaded and stored to computer, which were further used for docking process.
Molecular docking using exome horizon: Molecular docking is a key tool in structural molecular biology and computer assisting drug design. The goal of protein ligand docking is to predict the predominant binding mode(s) of a ligand with a protein of known 3D structure. Molecular docking between Drugs and PB2 protein 3D structure was done using EXOME Horizon (automated docking software by Mascon Global LTD). EXOME Horizon is an integrated system for bio-informatics. It has bio-informatics software suites required by high-end biological research labs. From suites for large genome analysis, to automated drug design, EXOME-Horizon is the one stop solution for all bio-computing needs. The package combines the robust Linux platform with customized, high performance hardware to facilitate efficient analysis of large amounts of data. Molecular docking followed by the preparation of ligand and receptor and finally docking the ligand with receptor has been performed. Receptor preparation has been done by removal of solvent molecules and removal of other unnecessary components like cofactors. In process of ligand preparation Simplified Molecular Input Line Entry System (SMILES) notation of drug was obtained from Drugbank database. EXOME Horizon was employed for further ligand preparatory steps, docking calculation and initial addition of hydrogen and cleaning the geometry of target molecule.
RESULTS AND DISCUSSION
The PB2 protein sequence of polymerase PB2 (A/chicken/India/WB-NIV529/2008(H5N1)) was retrieved from NCBI with Accession No. ACZ58135 and length 759 amino acids. When 3D structure of PB2 protein was searched, it was observed that no known 3D structure was available. Therefore the protein sequence was further used to perform homology modeling using Swiss model for template selection. A template (PDB ID: 2vqzD) was selected. This show a highest sequence homology of 92.727%, with atomic resolution of its X-ray crystal structure being 2.3 A and E-value being 6.91205e-84 (Fig. 1).
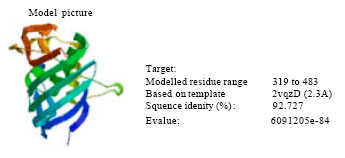 | |
| Fig. 1: | Homology Modeling Result obtained using Swiss Model. (Obtained from results of Swiss model) |
| Table 1: | Docking results obtained with free energies, active sites name and active site residues |
 | |
The predicted model was also checked for psi and phi torsion angles using the Ramchandran plots. The molecular visualization program Discovery Studio Visualizer (Accelrys) was used to manipulate the models based on residue interactions, energy minimization and steric hinderance. The model predicted by Swiss model was used for further analysis by PROCHECK (Laskowski et al., 1996). Ramchandran plot analysis (Fig. 2) shows 87.1% of the residues in the most favored region, 12.2% in the additional allowed, 0.7% in the generously allowed regions and 0.0% in the disallowed region.
EXOME Horizon was used for ligands preparation of all three drugs namely Rimantadine (Fig. 3), Amantadine and Zanamivir that were obtained using Drug Data Bank. Receptor preparation from 3D model structure of virus protein PB2 was also performed (Fig. 4). Molecular docking between receptor and all three ligands were performed. Docking result for ligand Rimantadine has been shown in Fig. 5. Similarly the results for rest of the two ligands were also obtained. Results were obtained along with their binding energies and their active sites. Residues which lie within 5 Angstrom unit area of ligand that interact with it through their side chain were identified and were considered as Active site residues.
Active sites contain 12 amino acid residues involved in docking (Table 1). From the results it was observed that Rimantadine is the best inhibitor for virus protein PB2.
The H5N1 sub-type is the only highly pathogenic avian viral sub-type that has been documented to cause an outbreak of respiratory disease in humans. The pathogenicity of avian H5N1 influenza viruses to mammals has been evolving since the mid-1980s (Chen et al., 2004). During the past 6 years, infection of humans with avian influenza viruses of three subtypes (H5, H7 and H9) has been detected on multiple occasions (Subbarao and Katz, 2000). In 1997, H5N1 avian influenza viruses transmitted from birds to humans in Hong Kong caused the deaths of 6 of 18 infected persons (Claas et al., 1998). Moreover, in 2003, antigenically and biologically novel H5N1 sub-type of influenza virus killed one of two infected humans (Sturm-Ramirez et al., 2004). Due to the rapid spread of the H5N1 virus, there is a growing concern that the disease could develop into a global human pandemic with the potential to kill people in millions.
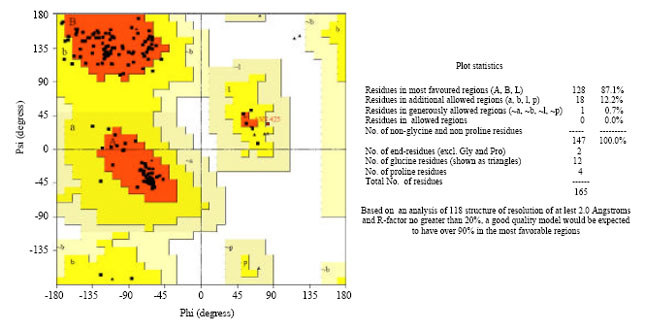 | |
| Fig. 2: | Ramachandran Plot analysis of predicted model using PROCHEK (Obtained from results of PROCHEK using SAVES) |
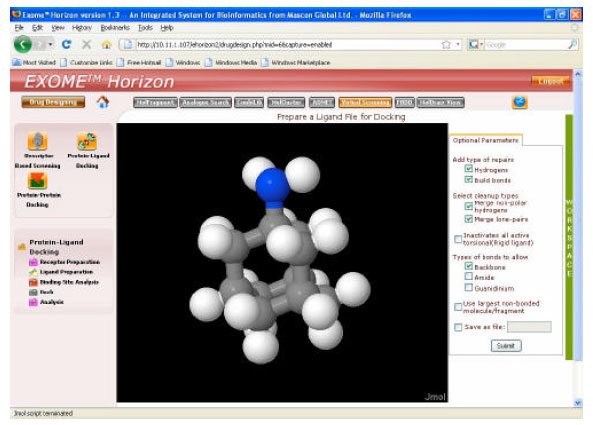 | |
| Fig. 3: | Ligand preparation of drug RIMANTADINE. (Exome Horizon, Meerut Institute of Engineering and Technology) |
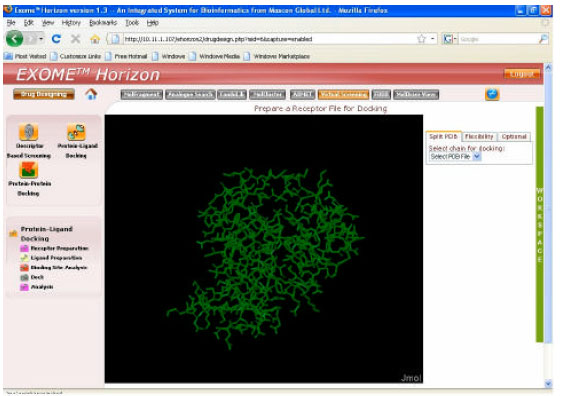 | |
| Fig. 4: | Receptor preparation. (Exome Horizon, Meerut Institute of Engineering and Technology) |
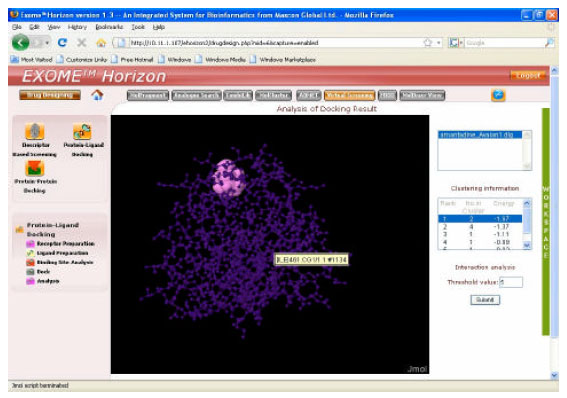 | |
| Fig. 5: | Docking of Receptor with the ligand RIMANTADINE. (Exome Horizon, Meerut Institute of Engineering and Technology) |
As at December 4, 2007, there were 336 confirmed human cases of H5N1 reported to WHO from different countries including Azerbaijan, Cambodia, china, Egypt, Indonesia, Iraq, Laos, Nigeria, Thailand, Turkey and Vietnam; of these cases, 207 (61.6%) people had died (Bello et al., 2008).
The genome of influenza viruses have 8 RNA segments encoding 10 proteins including two surface glycoprotein, haemaggulutinin (HA), neuraminidase (NA), nucleoproteins (NP), three polymerase proteins (PA,PB1,PB2) two matrix (M1,M2) and nonstructural proteins (NS1,NS2). The polymerase protein PB2 is 759 residues long influenza-A viruses. The polymerase complex is formed protein subunit interactions with the PB1 subunit (Digard et al., 1989). PB2 interacts with NP and not with PA (Biswas et al., 1998; Zurcher et al., 1996). This subunit plays an important role in transcription of mRNA by binding with the 5’ methylated cap of pre-mRNA in host cells for providing primers to viral mRNA synthesis (Nakagawa et al., 1995).
No 3D structure of PB2 protein was available in databases therefore a 3D model for PB2 protein was build based on the template (PDB ID: 2vqzD) with 92.727% sequence identity. The 3D model of PB2 protein has been evaluated and validated using Discovery Studio Visualizer (Accelrys) and PROCHECK. Ramchandran plot analysis shows the total number of residues in selected model was 165, with 128 (87.1%) residues in the most favored regions, 18 (12.2%) in the additional allowed, 1 (0.7%) in generously allowed region and 0 (0.0%) in the disallowed region.
It is widely accepted that drug activity is obtained through the molecular binding of one molecule (the ligand) to the pocket of another, usually larger, molecule (the receptor), which is commonly a protein. In their binding conformations, the molecules exhibit geometric and chemical complementarity, both of which are essential for successful drug activity. Therefore the model was used to create a receptor for molecular docking with the ligand of drugs rimantadine, amantadine, zanamivir using in silico methods. Results obtained after docking gives us free energies and active sites where the ligand binds to the receptor (Table 1). Rimantadine, having the least binding energy among the three drugs, is the most effective inhibitor for Virus protein PB2. The finding are unique in nature as no report is available about the homology modeling of PB2 and its docking with known drugs.
CONCLUSIONS
Prediction of protein 3D structure and its molecular docking with different ligands, with respect to any disease helps us to find out important aspects of protein structures such as their actives sites, binding energies etc. These in silico techniques help us to fulfill the initial phases of drug discovery and assist to extract the important information related to genes and proteins.
Here, we propose an idea that difference in binding energies might arise due to mutations which can cause conformational changes that affect the binding affinity of a ligand, as it is known that binding energies reflects the binding affinity . However, along with binding energies, several other physical effects like electrostatics, van der waals forces, hydrogen bonding and hydrophobic and entropic effects influence the binding affinity; these are also needed to be evaluated for calculating binding affinity of ligands or drug candidates.
The PB2 subunit plays an important role in transcription regulation in viral mRNA synthesis (Digard et al., 1989). In the present article we predicted how in silico analysis can be used for predicting the 3D structure of a target protein as well as checking the drug efficacy. Molecular docking and homology modeling of protein structure is the most important techniques to fulfill the desired result. These findings have implications in understating most specific drug for influenza virus.
REFERENCES
- Ahn, I., B.J. Jeong, S.E. Bae, J. Jung and H.S. Son, 2006. Genomic analysis of influenza A viruses, including avian flu (H5N1) strains. Eur. J. Epidemiol., 21: 511-519.
PubMedDirect Link - Anwar, T., S.K. Lal and A.U. Khan, 2006. In silico analysis of genes nucleoprotein, neuraminidase and hemagglutinin: A comparative study on different strains of influenza A (Bird flu) virus sub-type H5N1. In silico Biol., 6: 161-168.
Direct Link - Bello, M., B.M. Lukshi and M. Sanusi, 2008. Outbreaks of highly pathogenic avian influenza (H5N1) in Bauchi state, Nigeria. Int. J. Poult. Sci., 7: 450-456.
CrossRefDirect Link - Biswas, S.K., P.L. Boutz and D.P. Nayak, 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Gen. Virol., 72: 5493-5501.
Direct Link - Chen, H., G. Deng, Z. Li, G. Tian and Y. Li et al., 2004. The evolution of H5N1 influenza viruses in ducks in southern China. Proc. Natl. Acad. Sci., 101: 10452-10457.
CrossRef - Claas, E.C.J., A.D.M. Osterhaus, R. van Beek, J. de Jong and G.F. Rimmelzwaan et al., 1998. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet, 351: 472-477.
Direct Link - Digard, P., V.C. Blok and S.C. Inglis, 1989. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology, 171: 162-169.
PubMedDirect Link - Epstein, S.L., T.M. Tumpey, J.A. Misplon, C.Y. Lo and L.A. Cooper et al., 2002. DNA vaccine expressing conserved influenza virus proteins protective against H5N1 challenge infection in mice. Emerg. Infect. Dis., 8: 796-801.
PubMedDirect Link - Gasteiger, E., C. Hoogland, A. Gattiker, S. Duvaud, M.R. Wilkins, R.D. Appel and A. Bairoch, 2005. Protein Identification and Analysis Tools on the ExPASy Server. In: The Proteomics Protocols Handbook, Walker, J.M. (Ed.). 1st Edn., Humana Press, New Jersey, USA., ISBN-13: 978-1588295934, pp: 571-607.
- Honda, A., K. Mizumoto and A. Ishihama, 1999. Two separate sequences of PB2 subunit constitute the RNA cap-binding site of influenza virus RNA polymerase. Genes Cells, 4: 475-485.
PubMedDirect Link - Kitchen, D.B., H. Decornez, J.R. Furr and J. Bajorath, 2004. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discovery, 3: 935-949.
CrossRefDirect Link - Laskowski, R.A., J.A. Rullmann, M.W. MacArthur, R. Kaptein and J.M. Thornton, 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR., 8: 477-486.
CrossRefPubMedDirect Link - Marti-Renom, M.A., A.C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, 2000. Comparative protein structure modeling of genes and genomes. Ann. Rev. Biophys. Biomol. Struct., 29: 291-325.
CrossRefPubMedDirect Link - Nakagawa, Y., K. Oda and S. Nakada, 1995. The PB1 subunit alone can catalyze cRNA synthesis and the PA subunit in addition to the PB1 subunit is required for viral RNA synthesis in replication of the influenza virus genome. Virology, 70: 6390-6394.
PubMedDirect Link - Kamal, R.P., C. Tosh, B. Pattnaik, P. Behera and S. Nagarajan et al., 2007. Analysis of the PB2 gene reveals that Indian H5N1 influenza virus belongs to a mixed-migratory bird sub-lineage possessing the amino acid lysine at position 627 of the PB2 protein. Arch. Virol., 152: 1637-1644.
CrossRef - Ramachandran, G.N., C. Ramakrishnan and V. Sasisekharan, 1963. Stereochemistry of polypeptide chain configurations. J. Mol. Biol., 7: 95-99.
PubMedDirect Link - Sturm-Ramirez, K.M., T. Ellis, B. Bousfield, L. Bissett and K. Dyrting et al., 2004. Reemerging H5N1 influenza viruses in Hong Kong in 2002 are highly pathogenic to ducks. J. Virol., 78: 4892-4901.
Direct Link - Subbarao, K. and J. Katz, 2000. Avian influenza viruses infecting humans. Cell. Mol. Life Sci., 57: 1770-1784.
PubMed - Suzuki, Y. and M. Nei, 2002. Origin and Evolution of Influenza virus hemagglutinin genes. Mol. Biol. Evol., 19: 501-509.
PubMedDirect Link - Webster, R.G., W.J. Bean, O.T. Gorman, T.M. Chambers and Y. Kawaoka, 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev., 56: 152-179.
PubMedDirect Link - WHO, 2007. Update: WHO-confirmed human cases of avian influenza A (H5N1) infection, 25 November 2003-24 November 2006. Weekly Epidemiol. Rec., 82: 41-48.
Direct Link - Zurcher, T., S. De-la-Luna, J.J. Sanz-Ezquerro, A. Nieto and J. Ortin, 1996. Mutational analysis of the influenza virus A/Victoria/3/75 PA protein: Studies of interaction with PB1 protein and identification of a dominant negative mutant. J. Gen. Virol., 77: 1745-1749.
Direct Link