
Abdolkarim Sheikhi
Department of Immunology,
Yahya Jaberi
Department of Cardiology,
School of Medicine, Zanjan University of Medical Sciences, Zanjan, Iran
Abdolreza Esmaeilzadeh
Department of Immunology, School of Medicine,
Tarbiat Modarres University, Tehran, Iran
Mohammad Khani
Department of Cardiology,
School of Medicine, Zanjan University of Medical Sciences, Zanjan, Iran
Mehdi Moosaeefard
School of Nursing, Zanjan University of Medical Sciences, Zanjan, Iran
Mostafa Shafaqatian
Department of Immunology,
Pakistan Journal of Biological Sciences
Year: 2007 | Volume: 10 | Issue: 10 | Page No.: 1580-1587







ABSTRACT
Recent studies have shown that patients with heart failure over-express pro-inflammatory cytokines which enhance natural killer (NK) activity and negatively influence contractility and contribute to the remodeling of myocardium. The question is that how cardiovascular drugs influence on the cytokines of Peripheral Blood Mononuclear Cells (PBMCs) in Chronic Heart Failure (CHF). To study the effect of cardiovascular drugs on PBMCs-cytokines and NK activity of CHF patients. PBMCs of CHF patients/normal controls collected by Ficoll-paque density centrifugation. NK activity against K562 target cell was measured with MTT colorimetric assay. PBMCs were cultivated in RPMI/FCS, stimulated with phytohaemaglutinin (PHA). Tumor necrosis factor (TNF)-α interleukin (IL)-6, IL-2 and IL-1β of culture supernatants after 24 h incubation with/without furosemide, captopril and digoxin were measured with sandwitch ELISA. Patients had higher NK activity than controls (56.9% ±1.6 vs 50.9% ±1.2, p<0.05). NK activity of patients who already consumed Captopril/Furosemide didn’t show difference with controls. Captopril (3, 1, 0.3 μg mL-1) and Furosemide (5, 2.5, 1.25 μg mL-1) caused a dose dependent inhibition in TNF-α compared with control (329±23, 427±15, 519±19 and 343±19, 430±14, respectively vs. 562±24 pg mL-1 p<0.05). Furosemide caused a dose dependent decrease in IL-6 (421±31, 534±33 vs. 662±41 pg mL-1 p<0.05). Captopril and Furosemide didn’t show any significant effect on IL-1β/IL-2. Digoxin had no significant effect on PBMCs-cytokines. These data suggest that the immunomodulatory effects of Captopril and Furosemide may contribute to their beneficial and no long term adverse effects on PBMCs.
PDF Abstract XML References Citation
How to cite this article
Abdolkarim Sheikhi, Yahya Jaberi, Abdolreza Esmaeilzadeh, Mohammad Khani, Mehdi Moosaeefard and Mostafa Shafaqatian, 2007. The Effect of Cardiovascular Drugs on Pro-Inflammatory Cytokine Secretion
and Natural Killer Activity of Peripheral Blood Mononuclear Cells of Patients
with Chronic Heart Failure in vitro. Pakistan Journal of Biological Sciences, 10: 1580-1587.
DOI: 10.3923/pjbs.2007.1580.1587
URL: https://scialert.net/abstract/?doi=pjbs.2007.1580.1587
DOI: 10.3923/pjbs.2007.1580.1587
URL: https://scialert.net/abstract/?doi=pjbs.2007.1580.1587
INTRODUCTION
Chronic Heart Failure (CHF) is a complex syndrome that can result from any structural or functional cardiac disorder that impairs the ability of the heart to function as a pump to support a physiological circulation and its prognosis is comparable to that of different malignant diseases. The incidence of CHF increases exponentially with advancing age in humans and is the most common cause of hospitalization for adults over the age of 65 years (Jovicic et al., 2006). Patients with heart failure may have a number of symptoms, the most common being breathlessness, fatigue, exercise intolerance and fluid retention (Clark, 2006).
Any condition that damages the heart can lead to heart failure. The most common cause is coronary heart disease, although hypertension often coexists. Such patients usually have obvious abnormalities of the systolic function of the left ventricle (Cowie et al., 1997; Fox et al., 2001).
Paradigms and treatment strategies for heart failure have changed during the last decade. Traditionally, patients with CHF are treated with diuretics (e.g., Furosemide), vasodilators (e.g., Captopril) and inotropic (e.g., Digoxin) drugs, resulting in improvement in functional status and symptoms, but with no decrease in long-term mortality (Katz, 2003). The initial haemodynamic dysfunction of CHF has downstream effects on cardiovascular reflexes and systemic organ perfusion and function. The arterial under-filling is sensed by baroreceptors that activate powerful neurohormones, which act as effectors of vasoconstriction and of avid sodium and water retention. Recognition of neurohormones as important substances in the pathogenesis of CHF has resulted in several new treatment modalities, including angiotensin-converting enzyme (ACE) inhibitors, aldosterone antagonists and β-blockers, which yield marked improvements in morbidity and mortality of CHF patients (Packer et al., 1996 Medzhitov and Janeway, 2000). But CHF is still a progressive disease with high morbidity and mortality, suggesting that important pathogenic mechanisms remain active and unmodified by the present treatment modalities. Persistent immune activation and inflammation may represent such ‘unmodified mechanisms’. Attention has focused on mediators that are classically associated with innate immunity, including pro-inflammatory cytokines (von Haehling et al., 2004) and NK cells.
Cytokines are peptides that mediate cell-to-cell interactions via specific cell-surface receptors. They regulate activation, differentiation, growth, death and acquisition of effector functions of various cell types (So et al., 2006). They are increasingly recognized as important factors in the pathophysiology of CHF. The most important pro-inflammatory cytokines involved in CHF are TNF-α, IL-1β, IL-6 and IL-2.
The problem is that if traditional cardiovascular medicines are able to modify the pro-inflammatory cytokines and NK activity of PBMCs which are as important mediators in the pathophysiology of CHF.
In this study the question is that how traditional cardiovascular drugs (diuretics, angiotensin-converting enzyme (ACE) inhibitors and inotropics) influence on the overall cytokine network of PBMCs in CHF. The general objective of this research project is in vitro study of the effect of Captopril, Furosemide and Digoxin (as angiotensin-converting enzyme (ACE) inhibitor, diuretic and inotropic drugs, respectively) on cytokine secretion of PBMCs and measurement of NK activity of CHF patients before/after cardiovascular therapy. We hypothesize that each of above cardiovascular drugs alone or with together (mixed) might have inhibitory effect on cytokine secretion from cultured PBMCs. Also we hypothesize that patients who have consumed the drugs, would have lower NK activity.
MATERIALS AND METHODS
Patients: A total of 35 hospitalized (Dr. Beheshti hospital, Zanjan university of Medical Sciences, Zanjan, Iran) patients aged 53-84 years (65±13 year) were studied from 2005 to 2006. Among them 20 were males and 15 females. The spectrum of diseases were coronary artery disease (n = 35, all the patients), hypertension (n = 11), valvular heart disease (n = 12) and idiopathic dilated cardiomyopathy (n = 1). The diagnosis of CHF was based on a history of dyspnea and symptomatic exercise intolerance with signs of pulmonary congestion or peripheral edema, clinical examination and usual investigation. Among all subjects, 14 patients had New York Heart Association (NYHA) functional class II, 11 patients had NYHA functional class III and 10 patients had NYHA functional class IV. The patients with significantly concomitant disease such as chronic infectious, chronic obstructive pulmonary disease, cancer, diabetes mellitus, pregnancy, severe liver disease or connective tissue diseases were excluded. Patients with acute myocardial infarction, acute infectious or inflammatory diseases were also excluded, as was the use of drugs influencing immunoreactivity (i.e., corticosteroids). All subjects gave fully informed consent before taking 10 mL peripheral blood.
Preparation of human peripheral blood mononuclear cells: PBMC were obtained from CHF patients and collected by Ficoll-paque (Pharmacia) density centrifugation. The cells were washed three times with PBS, resuspended in RPMI-1640 media (Gibco), supplemented with 10% heat-inactivated fetal calf serum (Gibco), 100 μ mL-1 penicillin, 100 μg mL-1 streptomycin (Gibco) and cultured at 37°C in a humidified 5% CO2 atmosphere. The PBMCs were stimulated with phytohaemaglutinin (PHA; sigma) 10 μg mL-1 72 h then were washed 3 times before incubation with cardiovascular medicines.
NK activity assay
Preparation of target cell: The target cell was an erythroleukemia cell line named K562 a standard target cell for NK activity assay (Iran cell bank, Pastuer Institute, Tehran). The target cell was de-frozen and after three times washing, it was suspended in culture medium consisted of RPMI-1640 supplemented with 20% fetal calf serum (Gibco) and 100 μ mL-1 penicillin 100 μg mL-1 streptomycin.
Cytotoxicity assay: NK activity of PBMCs of CHF patients and normal controls were examined by means of a standard (4,5-dimethylthiazoyl-2,5-diphenyltetrazolium bromide; MTT; Sigma) assay using K562 cell line as the target. Cells were cultured at the effector/target (E/T) ratios of 2/1 and 0.5/1 in triplicate. The number of target cells in each well was 5x104. After the incubation period, 10 μL of MTT labeling reagent (0.5 mg mL-1) was added to each well. The plate was incubated for 4 h and then 100 μL of acidified isopropanol was added to the wells. After generation of the purple formazan crystals, absorbance of the samples was measured using an ELISA reader (Awareness, USA) at 570 nm and 630 nm as the reference wavelength.
Effect of cardiovascular medicines on PHA pre-stimulated PBMCs: PBMCs (2x106 cells mL-1 in 24-well plates) were stimulated with PHA (final concentration, 10 μg mL-1). After incubation of the 3 times washed PHA pre-stimulated PBMCs with captopril, furosemide or digoxin for 24 h, the supernatants were harvested and stored at-80°C until performance of the cytokine assay. 3 different concentrations of each drug (captopril 3, 1, 0.3 μg mL-1, furosemide 5, 2.5, 1.25 μg mL-1 and digoxin 3, 1.5, 0.75 ng mL-1) were tested separately. The effect of a mixture of the drugs with the above mentioned mead concentration was studied as well. The mead one was equal to therapeutic or normal plasma concentration of drug.
Cytokine assay: Cytokine concentrations in culture supernatants were determined by sandwich enzyme-linked immunosorbent assay (sandwich ELISA, Bendermed, Austria) according to the manufacturer’s instructions. A standard curve was generated with each set of samples assayed. Samples were frozen and thawed only once. All determinations were made in duplicate.
Statistical analysis: All values are presented as the mean±SE. The comparisons of the data of the effect of captopril, furosemide and digoxin on PHA pre-stimulated PBMCs-cytokine production was performed by Student’s t-test. Data were analyzed with SPSS (version 8.0). Statistical significance was assumed at a two-tailed value of p<0.05.
RESULTS
NK activity of 35 CHF patients (65±13 years, 20 patients after and 15 patients before consumption of Captopril/Furosemide) and 30 sex and age-matched normal controls was measured. As it is shown in Fig. 1, CHF patients had higher NK activity than normal controls (Mean±SE 56.9% ±1.6 vs 50.9%±1.2, respectively, p<0.05). Figure 2 indicates that NK activity of patients who already consumed Captopril and Furosemide didn’t show significant difference with normal controls.
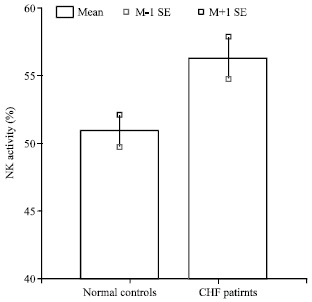 | |
| Fig. 1: | Natural killer (NK) activity of PBMCs of CHF patients and normal controls was measured against K562 target cell line with MTT colorimetric assay. NK activity of CHF patients (untreated and treated with together) was significantly higher than age-matched normal controls (p<0.05). Data are based on mean±standard error (SE) |
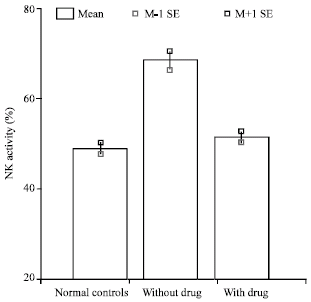 | |
| Fig. 2: | Natural killer (NK) activity of PBMCs of CHF patients and normal controls was measured against K562 target cell line with MTT colorimetric assay. NK activity of CHF patients after consumption of Captopril and Furosemide (treated group) doesn’t show any difference with age-matched normal controls. But patients before consumption of Captopril and Furosemide (untreated group) show higher NK activity compared with normal controls (p<0.05). Data are based on mean±standard error (SE) |
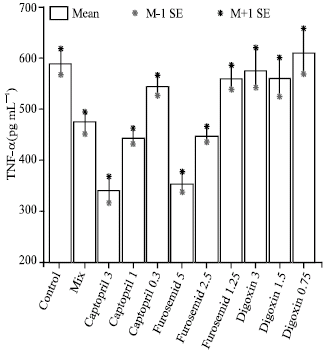 | |
| Fig. 3: | PHA-prestimulated PBMCs were incubated with 3, 1, 0.3 μg mL-1 Captopril, 5, 2.5, 1.25 μg mL-1 Furosemide, 3, 1.5, 0.75 ng mL-1 Digoxin and a mixture of Furosemide 2.5 μg mL-1, Captopril 1 μg mL-1 and Digoxin 1.5 ng mL-1 for 24 h. The supernatant of cultured PBMCs was used for cytokine assay with sandwich ELISA method. The PBMCs were incubated with PBS as control. Captopril and Furosemide caused a dose dependent inhibition but Digoxin did not show any effect on TNF-α secretion. A mixture of the above drugs had a significant inhibitory effect on TNF-α secretion as well. Data are based on mean±standard error (SE) |
As Fig. 3 shows, Captopril (3, 1, 0.3 μg mL-1) and Furosemide (5, 2.5, 1.25 μg mL-1) caused a dose dependent significant inhibition in TNF-α in comparison with control (329±23, 427±15, 519±19 and 343±19, 430±14, respectively vs 562±24 pg mL-1 p<0.05). A mixture of Captopril (1 μg mL-1), Furosemide (2.5 μg mL-1) and Digoxin (1.5 ng mL-1) caused a significant inhibition in TNF-α and IL-6 in comparison with control (447±103, 435±149, respectively vs 562±24 pg mL-1 p<0.05).
Figure 4 indicates that furosemide caused a dose dependent decrease in IL-6 (421±31, 534±33 vs 662±41 pg mL-1 p<0.05) but captopril had no effect on IL-6 secretion (647±177, 661±187, 674±182 pg mL-1).
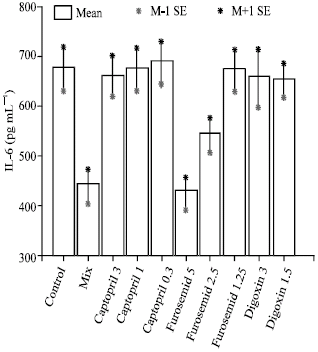 | |
| Fig. 4: | PHA-prestimulated PBMCs were incubated with 3, 1, 0.3 μg mL-1 Captopril, 5, 2.5, 1.25 μg mL-1 Furosemide, 3, 1.5, 0.75 ng mL-1 Digoxin and a mixture of Furosemide 2.5 μg mL-1, Captopril 1 μg mL-1 and Digoxin 1.5 ng mL-1 for 24 h. The supernatant of cultured PBMCs was used for cytokine assay with sandwich ELISA method. Furosemide caused a dose dependent inhibition but Digoxin and Captopril did not show any effect on IL-6 secretion. A mixture of the above drugs had a significant inhibitory effect on IL-6 secretion as well. The PBMCs was incubated with PBS as control. Data are based on mean±standard error (SE) |
Captopril and Furosemide didn’t show any significant effect on IL-1β and IL-2 (Fig. 5 and 6). Digoxin had no significant effect on cytokine secretion by PBMCs (Fig. 3-6).
The heart function classified by NYHA criteria (functional class) was correlated to concentrations of PBMC-TNF-α control/with Captopril and Furosemide. Patients with higher functional class have significantly higher TNF-α secretion (Table 1).
| Table 1: | PBMCs of patients with higher functional class produce higher TNF-α |
 | |
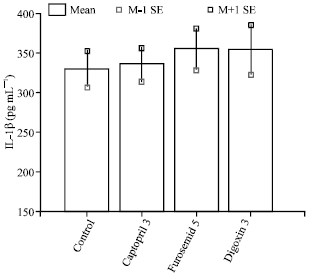 | |
| Fig. 5: | PHA-prestimulated PBMCs were incubated with 3 μg mL-1 Captopril, 5 μg mL-1 Furosemide, 3 ng mL-1 Digoxin for 24h. The supernatant of cultured PBMCs was used for cytokine assay with sandwich ELISA method. Captopril, Furosemide and Digoxin did not show any effect on IL-1β secretion. The PBMCs was incubated with PBS as control. Data are based on mean±standard error (SE) |
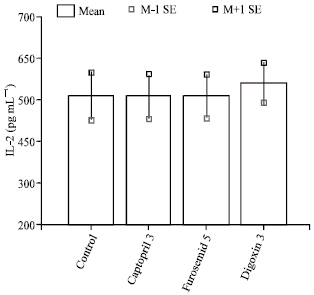 | |
| Fig. 6: | PHA-prestimulated PBMCs were incubated with 3 μg mL-1 captopril, 5 μg mL-1 furosemide, 3 ng mL-1 Digoxin for 24h. The supernatant of cultured PBMCs was used for cytokine assay with sandwich ELISA method. Captopril, Furosemide and Digoxin did not show any effect on IL-2 secretion. The PBMCs was incubated with PBS as control. Data are based on mean±standard error (SE) |
DISCUSSION
Previous studies have shown that some cardiovascular drugs have short-term beneficial effects on CHF such as traditionally treatment of CHF patients with diuretics, vasodilators and inotropic drugs, resulting in improvement in functional status and symptoms, but with no decrease in long-term mortality (Katz, 2003).
In the other hand, several studies have demonstrated that CHF patients are characterized by persistent immune activation in vivo. This is reflected in increased circulating levels of pro-inflammatory cytokines; TNF-α, IL-1β and IL-6 within the failing myocardium, independent of the cause of CHF (Torre-Amione et al., 1996; Straburzynska-Migaj et al., 2005; Damas et al., 2000). Although there is a lack of specificity of cytokine activation in CHF patients, several lines of evidence suggest that these inflammatory mediators are not only markers of immune activation, but may also play a pathogenic role in CHF. The pathogenic role of inflammatory cytokines in CHF is supported by research conducted in mouse models. Transgenic mice with cardiac over-expression of TNF-α developed dilated cardiomyopathy (Kubota et al., 1997). Also, systemic administration of TNF-α, even at concentrations comparable to those found in the circulation of CHF patients, have been shown to induce a dilated-cardiomyopathy-like phenotype in animal models (Bozkurt et al., 1998).
Inflammatory cytokines may modulate cardiovascular functions by a variety of mechanisms. Cytokines such as TNF-α and IL-1β have been shown to depress myocardial contractility. This may be due to uncoupling of β-adrenergic signaling, increase in cardiac nitric oxide, or alterations in intracellular calcium homeostasis (Yokoyama et al., 1993; El Sherif et al., 2005 and Goldhaber et al., 1996). TNF-α and members of the IL-6 family, may also induce structural changes in the failing myocardium such as cardiomyocyte hypertrophy and interstitial fibrosis (Yokoyama et al., 1997; Hirota et al., 1999). Additionally, TNF-α and IL-1β may promote cardiomyocyte apoptosis as well as activate metalloproteinases and impair the expression of their inhibitors, possibly contributing to cardiac remodeling (Krown et al., 1996; Pulkki 1997; Li et al., 2000). Experimental and clinical data have shown that circulating IL-2 is actively implicated in the pathophysiology of viral myocarditis and idiopathic dilated cardiomyopathy, reflecting an abnormal T-lymphocyte activation in these disorders (Nishio et al., 1999, Marriot et al., 1996).
In the present study the question was that how traditional cardiovascular drugs may change (if any) secretion of pro-inflammatory cytokine by PBMCs of CHF patients. Cultured PBMCs secrete cytokines in the supernatant but almost concentration of them is not detectable. Therefore we studied the effect of 3 different concentrations (the mead one of the 3 concentrations was equal to the normal plasma or therapeutic concentration) of the drugs on cytokine secretion by PHA pre-stimulated PBMCs. Captopril caused a dose dependent inhibition of TNF-α significantly but it doesn’t change IL-6, IL-1β and IL-2. The inhibitory effects of Captopril on production of pro-inflammatory cytokines have been also observed by others (Schindler et al., 1995, Peeters et al., 1998).
One of the major consequences of reduced cardiac performance in CHF is activation of the sympathetic nervous system and the rennin angiotensin system. Angiotensin II has chemotactic properties, binds to monocyte/macrophage and induces the up-regulation of TNF-α. The activation of immune system may account for the elevation of TNF-α secreted by PBMCs in patients with CHF. Angiotensin Converting Enzyme Inhibitors (ACEI) like Captopril are currently used as standard medical therapy in the management of CHF. Because it reduces angiotensin II, it is possible that ACEIs also has an effect on the anti-inflammatory cytokine TNF-α. The data presented in this study showed that the TNF-α production by PBMCs in patients with CHF was markedly decreased by Captopril. But the inhibition of angiotensin II by Captopril is not the sole pathway that affects cytokine production, because other potent ACEI such as Ramipril, Perindopril and Lisinopril, do not influence cytokine production (Schindler et al., 1995). Another pathway by which Captopril affects cytokine production may be in its capacity to induce prostaglandins (Katayama et al., 1989), potent inhibitors of cytokine synthesis.
Some previous studies show (Schindler et al., 1995) an inhibitory effect of Captopril on PBMCs-IL-1 in vitro which is not in agree with our current study which didn’t show any decreasing or increasing IL-1 secretion by PBMCs.
In heart failure, sodium is retained by the kidneys despite increases in extracellular volume. There is activation of rennin secretion, which culminates in the production of angiotensin II, causing vasoconstriction and aldosterone secretion. These synergistically produce an increase in tubular re-absorption of sodium and water. Diuretics such as Furosemide are the mainstay of symptomatic treatment to remove excess extracellular fluid in heart failure. In our study Furosemide caused a dose dependent down-regulation of TNF-α and IL-6 significantly in comparison with control but didn’t change IL-1β and IL-2. In another similar study it was demonstrated that Furosemide has inhibitory effect on cytokine release from normal human PBMCs (Yuengsrigul and Chin, 1999) but the mechanism of inhibitory effect of Furosemide is still unclear. So, in this study we showed that though Furosemide is consumed as symptomatic treatment, it is able to down-regulate TNF-α and IL-6, the main mediators of inflammation in CHF.
Digoxin had no increasing or decreasing effect on cytokine secretion of PBMCs in this study. Digoxin directly increases the force of myocardial contraction in normal individuals as well as in patients with heart failure. Studies have shown that treatment with Digoxin improves quality of life for patients with CHF. Better exercise times have been documented on stress testing and the incidence of symptomatic exacerbations of CHF has decreased. Patients most likely to benefit are those with the most severe heart failure and those with atrial fibrillation. But however use of the drug does not increase overall life expectancy in patients with CHF (Ahmed et al., 2006). In fact some previous studies show that Digoxin elevate pro-inflammatory cytokines in serum (Matsumori et al., 1999).
Patients with higher functional class had higher amount of PBMCs-TNF-α that is in agree with previous studies (Zhao and Xie 2001; Koller-Strametz et al., 1998) and confirm again that pro-inflammatory cytokines are pathogenic factors of CHF.
A mixture of the above drugs caused a significant inhibition of TNF-α and IL-6. It seems that the effect of the mixture is because of the effect of Captopril and Furosemide. We didn’t find a similar work to study the effect of a mixture of drugs on PBMCs cytokine secretion. This data say these medicines that usually are prescribed with together have no enhancing effect on PBMCs pro-inflammatory cytokine secretion of CHF patients.
In agree with previous studies (Sasayama and Matsumori, 1996; Sasayama et al., 1996; Prichett et al., 1995; Kanda et al., 1992) our data showed that untreated patients have higher NK activity than normal controls but patients who consumed Captopril/Furosemide had lower NK activity close to normal controls.
Cytokines form a vast array of relatively low molecular weight, pharmacologically active proteins. These substances are secreted by different cell types for the purpose of altering either their own function (autocrine) or that of adjacent cells (paracrine). The most important cytokines implicated in the progression of CHF are TNF-α, IL-1 and IL-6. These cytokines share some of their major characteristics (redundancy) and all act in a pro-inflammatory sense. Several hypotheses have been suggested to describe the origin of immune activation in CHF. Although the production of pro-inflammatory cytokines has mostly been attributed to secretion by mononuclear cells, but the myocardium is another important source of pro-inflammatory cytokines (Vassali 1992; Kapadia et al., 1997; Tsutamoto et al., 1998). Therefore, though we demonstrated the inhibitory effect of Captopril and Furosemide on the pro-inflammatory cytokine secretion by PBMCs which may contribute to their beneficial and no long-term adverse effects on PBMCs of CHF patients but still we don’t know the direct effect of these drugs on the cytokine secretion by myocardial tissue.
Despite of the important role of pro-inflammatory cytokines in the pathogenesis of CHF, but because of pleiotropism and redundancy of cytokines, anti-cytokine therapy has proven unsuccessful in CHF patients by now (Conraads, 2006).
ACKNOWLEDGMENTS
This work was supported by the grant number 199 from Zanjan University of Medical Sciences (ZUMSc). We thank Dr. Moosavi-nasab for assistance to do statistical analyses.
REFERENCES
- Ahmed, A., M.W. Rich, J.L. Fleg, M.R. Zile and J.B. Young et al., 2006. Effects of digoxin on morbidity and mortality in diastolic heart failure: the ancillary digitalis investigation group trial. Circulation, 114: 397-403.
Direct Link - Bozkurt, B., S.B. Kribbs, F.J. Clubb Jr., L.H. Michael and V.V. Didenko et al., 1998. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats Circulation, 97: 1382-1391.
CrossRefDirect Link - Conraads, V., 2006. Pro-inflammatory cytokines and their receptors in chronic heart failure: Do they really matter. Acta. Cardiol., 61: 161-168.
Direct Link - Cowie, M.R., A. Mosterd, D.A. Wood, J.W. Deckers and P.A. Poole-Wilson et al., 1997. The epidemiology of heart failure. Eur. Heart. J., 18: 208-225.
Direct Link - Damas, J.K., H.G. Eiken, E. Oie, V. Bjerkeli and A.Yndestad et al., 2000. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc. Res., 47: 778-787.
Direct Link - El-Sherif, W.T., L.F. Tooney, A.R. Meki and A. Abdel Moneim, 2005. Proinflammatory cytokines, soluble Fas receptor, nitric oxide and angiotensin converting enzyme in congestive heart failure. Egypt. J. Immunol., 12: 39-48.
Direct Link - Fox, K.F., M.R. Cowie, D.A. Wood, A.J.S. Coats and S.G. Thompson et al., 2002. Coronary artery disease as the cause of incident heart failure in the population. Eur. Heart. J., 22: 228-236.
Direct Link - Goldhaber, J.I., K.H. Kim, P.D. Natterson, T. Lawrence, P. Yang and J.N. Weiss, 1996. Effects of TNF-alpha on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am. J. Physiol., 271: H1449-H1455.
Direct Link - Hirota, H., J. Chen, U.A. Betz, K. Rajewsky, Y. Gu, J. Jr. Ross, W. Muller and K.R. Chien, 1999. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell, 97: 189-198.
Direct Link - Jovicic, A., J.M. Holroyd-Leduc and S.E. Straus, 2006. Effects of self-management intervention on health outcomes of patients with heart failure: A systematic review of randomized controlled trials. BMC Cardiovasc Disord, 6: 43-43.
Direct Link - Kanda, T., T. Yokoyama, T. Suzuki and K. Murata, 1992. Functional abnormalities of circulating natural killer cell subpopulations in patients with dilated cardiomyopathy. Tohoku J. Exp. Med., 168: 529-537.
Direct Link - Kapadia, S., H. Oral, J. Lee, M. Nakano, G.E. Taffet and D.L. Mann, 1997. Hemodynamic regulation of tumor necrosis factor-gene and protein expression in adult feline myocardium. Circ. Res., 81: 187-195.
Direct Link - Katz, A.M., 2003. Heart failure: A hemodynamic disorder complicated by maladaptive proliferative responses. J. Cell. Mol. Med., 7: 1-10.
Direct Link - Koller-Strametz, J., R. Pacher, B. Frey, T. Kos, W. Woloszczuk and B. Stanek, 1998. Circulating tumor necrosis factor-alpha levels in chronic heart failure: Relation to its soluble receptor II, interleukin-6 and neurohumoral variables. J. Heart Lung Transplant., 17: 356-362.
Direct Link - Krown, K.A., M.T. Page, C. Nguyen, D. Zechner and V. Gutierrez et al., 1996. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. J. Clin. Invest., 98: 2854-2865.
Direct Link - Kubota, T., C.F. McTiernan, C.S. Frye, S.E. Slawson and B.H. Lemster et al., 1997. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circulation Res., 81: 627-635.
Direct Link - Li, Y.Y., Y.Q. Feng, T. Kadokami, C.F. McTiernan, R. Draviam, S.C. Watkins and A.M. Feldman, 2000. Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. Proc. Nat. Acad. Sci. USA., 97: 12746-12751.
Direct Link - Marriot, J.B., J.H. Goldman, P.J. Keeling, M.K. Baig, A.G. Dalgleish and W.J. McKenna, 1996. Abnormal cytokine profiles in patients with idiopathic dilated cardiomyopathy and their asymptomatic relatives. Heart, 75: 287-290.
Direct Link - Matsumori, A., H. Igata, K. Ono, A. Iwasaki and T. Miyamoto et al., 1999. High doses of digitalis increase the myocardial production of proinflammatory cytokines and worsen myocardial injury in viral myocarditis: A possible mechanism of digitalis toxicity. Jap. Circ. J., 63: 934-940.
Direct Link - Nishio R, A. Matsumori, T. Shioi, H. Ishida and S. Sasayama, 1999. Treatment of experimental viral myocarditis with interleukin-10. Circulation, 100: 1102-1108.
Direct Link - Packer, M., M.R. Bristow, J.N. Cohn, W.S. Colucci, M.B. Fowler, E.M. Gilbert and N.H. Shusterman, 1996. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N. Engl. J. Med., 334: 1349-1355.
Direct Link - Peeters, A.C.T.M., M.G. Netea, B.J. Kullberg, T. Thien J.W.M. and Van der Meer, 1998. The effect of renin-angiotensin system inhibitors on pro-and anti-inflammatory cytokine production. Immunology, 94: 376-379.
Direct Link - Prichett, G., H.J. Cohen, K.M. Rao, F. Cobb, M. Sulivan and M.S. Currie, 1995. TNF, NK activity and measures of immune functions and inflammation in elderly men with heart failure. Gerontology, 41: 45-56.
Direct Link - Sasayama, S. and A. Matsumori, 1996. Vesnarinone: A potential cytokine inhibitor. J. Card. Fail., 2: 251-258.
Direct Link - Sasayama, S., A. Matsumori, Y. Matoba, S. Matsui and T. Yamada et al., 1996. Immunomodulation: A new horizon for medical treatment of heart failure. J. Card. Fail., 2: S287-S294.
Direct Link - Schindler, R., C.A. Dinarello and K.M. Koch, 1995. Angiotensin-convert- ing-enzyme inhibitors suppress synthesis of tumour necrosis factor and interleukin 1 by human peripheral blood cells. Cytokine, 7: 526-533.
Direct Link - So, T., S.W. Lee and M. Croft, 2006. Tumor necrosis factor/tumor necrosis factor receptor family members that positively regulate immunity. Int. J. Hematol., 83: 1-11.
Direct Link - Straburzynska-Migaj, E., R. Ochotny, A. Wachowiak-Baszynska, A. Straburzynska-Lupa, K. Lesniewska, K. Wiktorowicz and A. Cieslinski, 2005. Cytokines and heart rate variability in patients with chronic heart failure. Kardiol. Pol., 63: 478-485.
Direct Link - Torre-Amione, G., S. Kapadia, J. Lee, J.B. Durand, R.D. Bies, J.B. Young and D.L. Mann, 1996. Tumor necrosis factor-α and tumor necrosis factor receptors in the failing human heart. Circulation, 93: 704-711.
Direct Link - Tsutamoto, T., T. Hisanaga, A. Wada, K. Maeda and M. Ohnishi et al., 1998. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure and high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol., 31: 391-398.
Direct Link - Vassalli, P., 1992. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol., 10: 411-452.
Direct Link - Von-Haehling, S., E.A. Jankowska and S.D. Anker, 2004. Tumour necrosis factor-alpha and the failing heart-pathophysiology and therapeutic implications. Basic. Res. Cardiol., 99: 18-28.
Direct Link - Yokoyama, T., M. Nakano, J.L. Bednarczyk, B.W. McIntyre, M. Entman and D.L. Mann, 1997. Tumor necrosis factor-alpha provokes a hyper- trophic growth response in adult cardiac myocytes. Circulation, 95: 1247-1252.
Direct Link - Yuengsrigul, A. and T.W. Chin, 1999. Nussbaum E. Immunosuppressive and cytotoxic effects of furosemide on human peripheral blood mononuclear cells. Ann. Allergy Asthma. Immunol., 83: 559-566.
Direct Link - Zhao, S.P. and X.M. Xie, 2001. Captopril inhibits the production of tumor necrosis factor-a by human mononuclear cells in patients with congestive heart failure. Clinica. Chimica. Acta., 304: 85-90.
Direct Link