
M. Qayyum Khan
Department of Plant Breeding and Genetics, University of Azad Jammu and Kashmir, Faculty of Agriculture, Rawalakot Azad Jammu and Kashmir, Pakistan
Mukhtar Alam
Pakistan Oil Development Board, Ministry of Food Agriculture and Livestock Islamabad, Pakistan
R.J. K. Narayan
Institute of Biological Science, University of Wales, Aberystwyth, UK
M. Ilyas Khan
Department of Plant Breeding and Genetics, University of Azad Jammu and Kashmir, Faculty of Agriculture, Rawalakot Azad Jammu and Kashmir, Pakistan
Pakistan Journal of Biological Sciences
Year: 2002 | Volume: 5 | Issue: 7 | Page No.: 780-783







ABSTRACT
Twenty six-mer forward and 23-mer reverse primers were designed using the published nucleotide sequence data of cellular T-DNA of Nicotiana glauca. Primers annealing temperature and Mg+2 concentration were optimized for a polymerase chain reaction. At target sequence of about 1050 bp overlapping the rolB and rolC genes were successfully amplified from the genomic DNA of N. glauca. The amplified sequence was cloned into pUC18 vector. The cloned rol B-C fragment showed a high base sequence homology to the rol B and C genes located on the T-DNA of Ti and Ri plasmids of Agrobacterium tumefaciens and Agrobacterium rhizogenes respectively.
PDF Abstract XML References Citation
How to cite this article
M. Qayyum Khan, Mukhtar Alam, R.J. K. Narayan and M. Ilyas Khan, 2002. PCR Amplification and Cloning of T-DNA Homologous Sequences from Nicotiana glauca. Pakistan Journal of Biological Sciences, 5: 780-783.
DOI: 10.3923/pjbs.2002.780.783
URL: https://scialert.net/abstract/?doi=pjbs.2002.780.783
DOI: 10.3923/pjbs.2002.780.783
URL: https://scialert.net/abstract/?doi=pjbs.2002.780.783
INTRODUCTION
Infection of dicotyledenous plants by the soil bacteria Agrobacterium tumefaciens and Agrobacterium rhizogenes induces neoplastic growth in the host plants (Watson et al., 1975). A unique feature of this bacteria-plant interaction is the transfer of a segment of bacterial genome into the plant genome and its integration there (Van Larbeke et al., 1974, 1975). The segment of DNA which is transferred into the host tissue is called transferred DNA (hence T-DNA) (Bevan and Chilton, 1982). T-DNA resides in the Ti (tumor inducing) or Ri (root inducing) plasmids of A. tumefaciens and A. rhizogenes respectively (Chilton et al., 1982). White et al. (1982, 1983) made an interesting discovery when they detected T-DNA sequences of homologous T-region of RiA4 plasmid in the genomic DNA of untransformed N. glauca plant. The Ri TL-DNA sequences in the plant genome were termed as cellular T-DNA or (cT-DNA) sequences. Furner et al. (1986) were able to confirm that the cT-DNA sequences were indeed homologous to a part of the T-region of Ri plasmid. In the genome of N. glauca, the cT-DNA sequences are organized in the form of an imperfect inverted repeat about 13kb length. The homology between cT-DNA and T-DNA of Ri plasmid is specially high in certain regions such as the 88% homology between the rooting loci of A. rhizogenes Ri plasmid (rol A, rol B and rol D) which are concerned with the hairy root induction in the infected plants. The corresponding sequences (designated Ng rol) even contain potential reading frames corresponding to their counter parts on the Ri plasmids.
Whether the cT-DNA is originally of eukaryotic or prokaryotic origin, Tepfer (1983) and White et al. (1983) maintain that probably in the evolution of Agrobacterium-plant relationships the bacterium has captured plant genes which it re-inserts into the plant genome. An alternate theory has been proposed by Furner et al. (1986) which holds that early in the evolution of genus Nicotiana an infection by the Agrobacterium resulted in the introduction of T-DNA into the plant genome. The interkingdom gene transfer in Agrobacterium-plant interaction and the uncertain origins and role of the cT-DNA makes it an interesting material from genetic engineering and evolutionary point of view.
The present investigation was carried out to device PCR based method for amplifying rolB and rolC gene sequences contained within the left arm of cT-DNA. This method will make their isolation and survey in other species highly efficient.
MATERIALS AND METHODS
Plants were grown at the green house facilities of IBS, University of Wales Aberystwyth, Dyfed UK.
DNA extraction: Two grams fresh leaves powdered material was suspended in 15ml of extraction buffer [100mM Tris-HCl (pH=8.0), 50mM EDTA, 0.5M NaCl, 2% (W/V) SDS and 1% (W/V) polyvenyl pyrrolidone (PVP)]. The extraction buffer/lysis buffer was pre-warmed to 65oC and β- mercaptoethanol was added up to a final concentration of 10mM just before use. The sample was vortexed briefly and then incubated at 65oC for 60 minutes in a water bath mixing once in every 10 minutes. Ten ml of chloroform-octanol (24:1 V/V) was added to the lysate and the contents of tubes were mixed by inverting 20-30 times. The tubes were centrifuged for 10 minutes at 10,000rpm at 4oC. The aqueous phase was transferred to a fresh tube and 2/3 volume of pre-cooled (-20oC) isopropanol was added to the tubes. Contents of the tubes were carefully mixed and incubated at -20oC for 2 hours. DNA precipitate was spooled out with a sterile glass rod or collected by centrifugation at 10,000rpm for 10 minutes at 4 oC. The pellet was washed with 70% ethanol, vacuum dried for 2-3 minutes and dissolved in TE buffer.
Plant DNA/plasmid DNA/ or PCR products were fractionated by agarose gel electrophoresis (2.5V/cm). DNA was transferred from agarose gel to nylon filters by capillary blotting as described in Sambrook et al. (1989). Radioactive labeling was done according to the procedure described by Feinberg and Vogelstein (1983). Hybridization and pre-hybridization procedures were the same as described by Sambrook et al. (1989). Autoradiograpghs were obtained by exposing the filters to X-rays film (Kodak X-omat AR). The exposure was carried out at -700C in a exposure cassette internally lined with intensifying cawo screen.
PCR amplification: PCR amplification was done as described by Mullis and Faloona, (1987). 10X PCR buffer II (500mM KCl, 100mM Tris- HCl, pH=8.0, 25mM MgCl2 and Taq DNA polymerase (5units/μl) were purchased from Advanced Biotechnologies (UK). A dNTPs mixture (1.25mM of each dATP, dCTP, dTTP and dGTP), forward and reverse primers (0.25μM each), sterile water (HPLC grade) and target DNA were also required. Five μl of 10X PCR buffer, 9μl of 25μl MgCl2 solution, 2.5μl of each dNTP mixture, 0.5μl of the forward and reverse primers and 100ng of the target DNA were mixed in 0.5ml tube for a typical 50μl reaction. Volume (including the enzyme) was made up to 50μl with HPLC grade distilled water. The contents of the tubes were gently mixed and briefly centrifuged. Two μl of Taq DNA polymerase enzyme were added to the tubes and mixed with a pipette tip. The reaction was overlaid with a 40μl of mineral oil. The PCR was carried out in the Thermal-cycler (Perkin- Elmer).
Elution of DNA was carried out by the procedure described by Heery et al. (1990). Cloning of the PCR amplified fragment was accomplished by using the Sure ClonTM ligation system supplied by Pharmacia according to the manufacturer instructions. The PCR
amplified fragment was ligated into the pUC18 vector. E. coli strain NM522 was transformed with this vector. Small scale preparation of E. coli plasmid DNA was done according to the procedure of Sambrook et al. (1989).
RESULTS AND DISCUSSION
a) Primer design: The cT-DNA sequences were first detected by White et al. (1982, 1985) in the genomes of certain Nicotiana species. However, detailed analysis of the nucleotide sequences and a comparison with the T-region of Ri and Ti plasmid was done only for the cT-DNA of N. glauca (Furner et al., 1986). PCR primers were designed from this published sequence data of the cT-DNA.
The computer software Oligo version 2. identified a 26-mer forward and 23-mer reverse primers with the following sequences:
Nfor 162 55’-TTTGTCGAAGTTAGCTCCATTCTGCTC-3’
Nrev 2761 5’-TAGGCTTCTTTCTTCAGGTTAC-3’
The target that would be amplified using these primers is a region of the cT-DNA overlapping the Ng rolB and Ng rolC genes. The amplified sequence will be referred to as cellular rolB-C sequences in this paper.
b) Optimization of Mg+2 concentration and primer annealing temperature: Mg+2 concentration and primer annealing temperature both effect the specificity of the amplification process and the activity of the Taq polymerase. For a specific amplification of the target DNA and for elimination of the primer-dimer artifacts on optimization of the two parameters was necessary. The effect of different levels of Mg+2 on the amplification process is given in Fig. 1. The primer annealing temperature at this stage was kept very low (42oC). The DNA thermal cycler was performed for the following parameters: 1 cycle 94oC , 4min; 35 cycles 94oC, 4min; 42oC 1min 30sec., 72oC 1min 30sec and 1 cycle 72oC, 10min. No amplification occurred in the absence of Mg+2 (Lane 2) (Fig. 1).
Lane 3-8 shows amplification of several bands including a prominent band of the expected size (≈ 1050 bp). It is clear that the band of the expected size appears most prominent (lane 4) when the amplification was carried out at Mg+2 level of 6mM final concentration.
Several non-specific fragments were amplified along with the target sequence when the primer annealing temperature was 42oC (Fig. 1). Therefore, more stringent annealing temperatures (45, 47 and 50oC) were used keeping Mg+2 at the optimized level. The non-specific amplification was eliminated at the annealing temperature of 45oC (Fig. 2). A single band of 1050bp is amplified which corresponds to the expected molecular weight. No amplification products were obtained at the higher annealing temperature of 47 and 50oC.
c) Cloning of rolB-C sequence amplified from the DNA of N. glauca: The PCR product was fractionated on 1% agarose gel and the desired fragment was eluted from it according to the procedure of Heery et al. (1990). The same was cloned into the smalI site of the pUC18 vector using the SureClone kit (Pharmecia Ltd.) according to the manufacturer instructions. Bacterial strain NM522 was transferred with the pUC 18 vector. Successful cloning was confirmed by blue/white selection recombinants colonies, antibiotic resistance of the transformed bacteria. Finally, the insert sequences contained within the pUC18 were PCR amplified using the M13 forward and reverse universal primers.
The PCR product obtained from the recumbent plasmid in lane 7 contains an insert having the expected size (Fig. 3). Lane 2-6 represents recombinants plasmids showing cloned sequences of relatively smaller molecular sizes.
d) Base sequence homology of the cloned rolB-C sequence to the rol B and rol C gene of A. rhizogenes: The rolB and rolC genes of A. rhizogene and A. tumefaciens are located inside the T-region of Ri and Ti plasmids respectively. It was necessary to confirm that the cloned cellular rolB-C fragment was homologous to the corresponding region in the Ri and Ti plasmids.
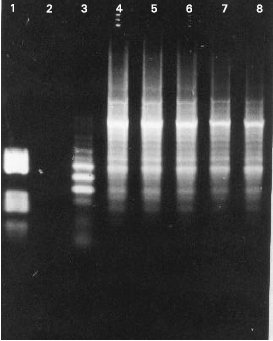 | |
| Fig. 1: | Optimization of Mg+2 level for PCR amplification of rolB-C sequences from the genomic DNA of N. glauca. Lane 1. contains marker DNA, Lane 2. No added Mg+2, Lane 3. 3mM, Lane 4. 6mM, Lane 5. 9mM, Lane 6. 12mM, Lane 7. 15mM and Lane 8. 18mM final concentration respectively. |
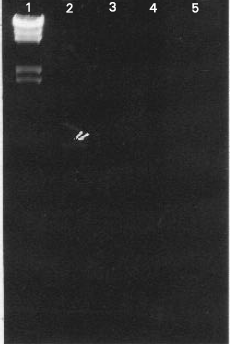 | |
| Fig. 2: | Optimization of annealing temperature for PCR amplification of rolB-C sequence from the genomic DNA of N. glauca. Lane 1 contains marker DNA. Lanes 2,3 and 4 contain PCR products obtained at annealing temperature of 45, 47 and 50°C respectively. Specific amplification was obtained in Lane 1. No amplification was observed at higher annealing temperatures. Amplification at lower temperature of 42. |
The rolB and rolC gene sequences from clones (provided by Dr. Khikawa Takanari, University of Tokyo) were fractionated in 1% agarose gel and
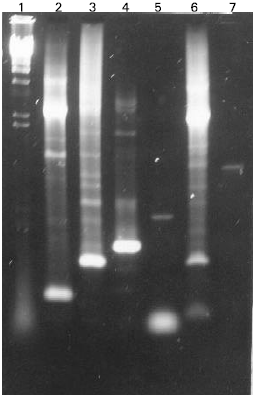 | |
| Fig. 3: | Cloning of rol-B-C sequences amplified from N. glauca DNA into pUC18 vector. PCR amplification of insert sequences in plasmid DNA of selected bacterial colonies. Lane 7 contains an insert having the expected size. Lane 2 to 6 represent recombinant plasmid, showing cloned sequences of relatively smaller molecular sizes. Lane 1 contains molecular size marker (lambda DNA Hind III) |
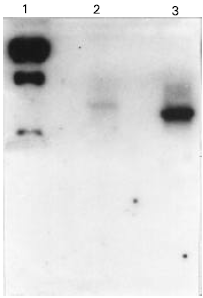 | |
| Fig. 4: | Autoradiography showing base sequence homology of the cloned cellular rol B-C sequence to the rolB and rolC genes of A. rhizogenes. rolB and rolC gene sequences were fractionated electrophoretically in an agarose gel and transferred to nylon filters. The cloned rol B-C sequences was radioactively labeled and used as a molecular probe for hybridization. Homology was detected between cellular rolB-C sequence and bacterial rolB (2) and rolC (3) gene sequences. Lane 1 end labeled lambda X Hind III |
immobilized on Nylon filters. The filters were probed with 32P labeled cellular rolB-C sequence amplified from the genomic DNA of N. glauca. The cellular rolB -C sequence possessed a high level of base sequence homology to rolB (Lane) and rolC (lane 3) genes of A. rhizogenes (Fig. 4). Since pre-hybridization and hybridization was carried out at a high temperature (65oC) and followed by stringent washing protocol therefore it can be concluded that the level of homology is substantially high. Homology to both rolB and rolC sequences is to be expected because the primers were designed for a region of cT -DNA that overlaps both the rolB and rolC genes.Furner et al., 1986 has reported specially a high homology for the rol gene loci between T-DNA of A. rhizogenes and cT-DNA of N. glauca.
Research into the molecular mechanism underlying gene transfer and the subsequent tumorigeneses is important because it illustrates a complex biological process of cancerous cell proliferation simplified form. Tumor induction by Agrobacterium species through the transfer of DNA represents a phenomenon which is parallel to the tumor induction by retroviruses (Gheysen et al., 1985). Several species of plants which are known to be amenable to transformation by Agrobacterium posses cT-DNA sequences. In contrast species which have consistently failed to be transformed such as Lathyrus, do not possess cT-DNA sequences. It is meaningful to wide the search for cT-DNA homologous sequences in diverse plant genera and find out whether there is any correlation between the presence of cT-DNA and the ability of a plant species to be transformed.
With a simplified PCR based techniques as detailed above, it should be possible to conduct a detailed survey of cT-DNA sequences in genera where evolutionary relationships between species included are already well understood. A link between genus Nicotiana and cT-DNA has been well established (White et al., 1983; Furner et al., 1986) and the evolution of the genus has been described in detailed (Goodspeed, 1954; Narayan, 1987).
In conclusion the cT-DNA of N. glauca possessed a high level of base sequence homology to rolB and rolC gene of A. rhizogenes. Our results also raise the possibility that how widely the rolB-C sequences are distributed among the subgeneric sections of genus Nicotiana. The pattern of distribution may have occurred through the repeated Agrobacterium-mediated DNA transfer of different stages in the evolution of the genus. So a detailed survey of the distribution of rolB-C sequences in the genus Nicotiana may indicate that the presence of rolB and rolC gene sequences in species of the genus pre-dates the divergence of Cestroid and Petuniod complexes, which represents the highly concerned regions of cT-DNA and T-DNA can furnish answer to questions such as: How did the Agrobacterium-plant relationship originate? and whether the interkingom gene transfer has influenced the evolution of plant and Agrobacterium species.
REFERENCES
- Feinberg, A.P. and B. Vogelstein, 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem., 132: 6-13.
CrossRefDirect Link - Furner, I.J., G.A. Huffman, R.M. Amasino, D.J. Garfinkel, M.P. Gordon and E.W. Nester, 1986. An Agrobacterium transformation in the evolution of the genus Nicotiana. Nature, 319: 422-427.
Direct Link - Mullis, K.B. and F.A. Faloona, 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol., 155: 335-350.
CrossRefPubMedDirect Link - White, F.F., B.H. Taylor, G.A. Huffman, M.P. Gordon and E.W. Nester, 1985. Molecular and genetic analysis of the transferred DNA regions of the root-inducing plasmid of Agrobacterium rhizogenes. J. Bacteriol., 164: 33-44.
PubMedDirect Link