
J.K. Ebigwai
Department of Plant and Ecological Studies, University of Calabar, Calabar, Nigeria
LiveDNA: 234.31468
F.A. Akomaye
Department of Plant and Ecological Studies, University of Calabar, Calabar, Nigeria
G.M. Ubi
Department of Genetics and Biotechnology, University of Calabar, Calabar, Nigeria
Journal of Biological Sciences
Year: 2020 | Volume: 20 | Issue: 1 | Page No.: 13-21







ABSTRACT
Background and Objective: Morphological mimicry resulting in nomenclatural ambiguities between and among plant taxa is fast becoming a major concern in natural product applications. Frequent nomenclatural substitution between Tetraptera tetraptera and Albizia adianthifolia caused by morphological similarities was resolved taxonomically using Maturase K gene as a DNA barcoding tool. Materials and Methods: Grounded leaf samples of the specimens were subjected to standard DNA protocols of extraction, amplification, sequencing and BLASTING (Basic Local Alignment Tool). Results: The results of differential nucleic acid purity levels, a sequence length of 755 and 766 base pairs respectively, presence of 728 conserved codon, 38 single nucleotide polymorphisms (SNP) between both samples and varied amino acid residues proved both specimens as distinct taxon. When the sequenced products were subjected to Basic Local Alignment Tool, specimen A exhibited a 99.2% homology with Tetrapleura tetraptera while specimen B presented a 99.7% homology with Albizia adianthifolia and Albizia petersiana indicating congruent identity and hence nomenclatural ambiguity. The phylogenic tree constructed to resolving the congruency revealed specimen B as exhibiting closer taxonomic distance to Albizia adianthifolia than Albizia petersiana. Conclusion: The study concludes by recommending DNA barcoding as a potent tool to resolving nomenclatural conflicts among morphological similar taxa.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
J.K. Ebigwai, F.A. Akomaye and G.M. Ubi, 2020. Resolving Taxonomic Ambiguity Between Two Morphological Similar Plant Taxa Using Maturase K Gene Analysis. Journal of Biological Sciences, 20: 13-21.
DOI: 10.3923/jbs.2020.13.21
URL: https://scialert.net/abstract/?doi=jbs.2020.13.21
DOI: 10.3923/jbs.2020.13.21
URL: https://scialert.net/abstract/?doi=jbs.2020.13.21
INTRODUCTION
Accurate plant nomenclatural identity is relevant to scientific1, agricultural2 and medical enterprise. It has remained the underlying denominator for conservation, pharmacological and bio stratigraphic practice. Utilization of wood and wood products for various ecosystem services have further added premium to the need for timely and accurate taxa identification3. Environmental mimicry, mutation and high rate of speciation had resulted in morphological ambiguities even among members of supposedly distant clades4. This had made traditional means of authentication such as expert recognition and species matching with voucher specimens almost unusable and untenable. The choice of any novel systematic tool to be applied in resolving nomenclatural opacities is centered on matching obtained information to its closest homologue in a data bank. This criterion often confines application of anatomical, cytological, paleontological, phytochemical and serological markers to restricted taxa5,6. The existence of gene banks in several regions of the world has made choice of molecular barcoding enticing and readily applicable.
Several DNA primers have been successfully employed in resolving taxonomic conflicts across all nomenclatural hierarchies. Ribulose bisphosphate carboxylase (Rbcl) has been applied in several studies7-9, were RubisCo was the choice primer10 and Maturase K gene was successfully used11-13. These primers are used primarily because of their reduced intraspecific variations.
Tetrapluera tetraptera and Albizia adianthifolia are two morphologically similar species whose nomenclatural identities are often confused with each other. Since they are reservoirs of varied chemical information, assigning inaccurate name could elicit reactions that could imperil lives. The aim of the research was to compare the sequenced Maturase k regions of the two morphological similar specimens with those in NCBI database in order to obtain their accurate nomenclature.
MATERIALS AND METHODS
Study area: Samples were collected within Calabar Metropolis, Nigeria in June, 2019. DNA extraction and quantification was carried out in the Molecular Biology laboratory of the Department of Genetics and Biotechnology, University of Calabar while Polymerase Chain Reaction (PCR) and Sequencing were done by Inqaba Biotech, Pretoria, South Africa. The analyses spanned between June-July, 2019.
 | |
| Fig. 1(a-b): | (a) Specimen A and (b) Specimen B |
Plant material collection: Fresh plant leaves were collected within University of Calabar, Calabar Cross River State. The samples were adequately authenticated to subfamily level using Kang et al.14 procedures. Field identification was based on folia, pods and inflorescence characteristics. Their affinity was further confirmed at the Herbarium unit of the Department of Plant and Ecological Studies, University of Calabar, Calabar, Nigeria (Fig. 1a, b).
DNA extraction: Extraction was done using a Zymo plant/seed DNA mini prep extraction kit. One hundred and fifty milligrams (150 mg) of the plant leaves were transferred into ZR Bashing Bead Lysis tubes, 750 μL of lysis solution were added to the tube. The tubes were secured in a bead beater fitted with a 2 mL tube holder assembly and processed at maximum speed for 5 min. The ZR bashing bead lysis tube was centrifuged at 10,000×g for 1 min. Four hundred microliters (400 μL) of supernatant was transferred to a Zymo-Spin IV spin Filter (orange top) in a collection tube and centrifuged at 7000×g for 1 min. One thousand two hundred microliters (1200 μL) of fungal/bacterial DNA binding buffer was added to the filtrate in the collection tubes bringing the final volume to 1600 μL, 800 μL was then transferred to a Zymo-Spin IIC column in a collection tube and centrifuged at 10,000×g for 1 min, the flow through was discarded from the collection tube. The remaining volume was transferred to the same Zymo-spin and spun. Two hundred (200) μL of the DNA Pre-Was buffer was added to the Zymo-spin IIC in a new collection tube and spun at 10,000× g for 1 min followed by the addition of 500 μL of fungal/bacterial DNA wash buffer and centrifuged at 10,000× g for 1 min. The Zymo-spin IIC column was transferred to a clean 1.5 μL centrifuge tube, 100 μL of DNA elution buffer was added to the column matrix and centrifuged at 10,000× g for 30 sec to elute the DNA. The eluted DNA was transferred into Zymo-spin IV-HRC column into a 1.5 mL tube and spun at 10,000× g for 1 min. The product was then stored at -20°C for PCR.
DNA quantification: The extracted genomic DNA was quantified using the Nano drop 1000 spectrophotometer. The software of the equipment was launched by double clicking on the Nano drop icon. The equipment was initialized with 2 μL of sterile distilled water and blanked using normal saline. Two microliters of the extracted DNA was loaded onto the lower pedestal, the upper pedestal was brought down to contact the extracted DNA on the lower pedestal. The DNA concentration was measured by clicking on the “measure” button.
DNA amplification: For amplification and sequencing of MatK gene, primer pair: MatK-1RKIM-f and MatK-3FKIM-r were used following the method of Little and Stevenson11-13. The DNA was amplified on an ABI 9700 Applied Bio systems thermal cycler at a final volume of 30 μL for 35 cycles. The PCR mix included: the X2 Dream Taq Master Mix supplied by Inqaba, South Africa (Taq polymerase, DNTPs, MgCl) and the primers at a concentration of 0.5 μM and 25 ng of the extracted DNA as template.
The PCR conditions were as follows: Initial denaturation, 95°C for 5 min, denaturation, 95°C for 30 sec, annealing, 55°C for 40 sec, extension, 72°C for 50 sec for 35 cycles and final extension, 72°C for 5 min. The product was resolved on a 1% agarose gel at 130 V for 25 min and visualized on a blue light Tran’s illuminator for a base pair product size. Each PCR reaction was repeated 3 times on each sample and examined by electrophoresis on 1% agarose gel, using DNA marker 1 kb ladder.
Amplicons sequencing and sequence analysis: The Amplicons were sequenced using the BigDye Terminator kit on a 3510 ABI sequencer by Inqaba Biotechnological, Pretoria South Africa. The sequencing was done at a final volume of 10 μL, the components included 0.25 μL BigDye® terminator v1.1/v3.1, 2.25 μL of 5×BigDye sequencing buffer, 10 μM Primer PCR primer and 2-10 ng PCR template per 100 bp. The sequencing conditions were as follows 32 cycles of 96°C for 10 sec, 55°C for 5 sec and 60°C for 4 min. The obtained sequences were edited using the bioinformatics algorithm Trace edit. The nucleotide sequences were aligned using ClustalW in MEGA715,16 to identify parsimony informative sites (Single Nucleotides Polymorphism-SNP).
Sequence identification: Sequence homology of the two specimens was detected using Basic Local Alignment Tool (BLAST) for highly similar sequences from the National Center for Biotechnology Information (NCBI) non-redundant nucleotide (nr/nt) database17. The query sequence was identified based on the percentage identity or similarity with a known sequence18. The sample was said to be correctly identified when the highest BLAST (%) identity of the query sequence was from the expected species or the species belonging to the expected subfamily; ambiguous identification means that the highest BLAST (%) identity for a query sequence was found to match several species of the study subfamily; incorrect identification means that the highest BLAST (%) identity of the query sequence was not from the expected subfamily19. Phylogenetic tree method was used to resolve any ambiguous identification by BLAST. The MatK sequences of other members of same genus were mined from NCBI in addition to those generated in this study. The genetic distances for the sequenced samples were equally evaluated. The inter specific and intra specific distances were computed using the Maximum Composite Likelihood model in MEGA720,21. The pairwise alignment of nucleotide sequence was done using ClustalW to identify any parsimony informative sites.
Statistical analysis: Data generated from the study were collated and subjected to statistical analysis using the analysis of variance (ANOVA) procedures of GENSTAT vr 12 of 2016 (https://library2.lincoln.ac.nz/documents/Genstat%20ANOVA%201%20workshop%20(16th%20ed).pdf) for morphological data. Sequence amplicons were edited using the Chromas software. The sequences were aligned and analyzed using the molecular evolutionary genetic analysis (MEGA) 7.0 software, Darwin 5.5 software and Bioedit software.
RESULTS
DNA quality: Result showed that the A260/280 samples A and B were 0.8 and 1.0, respectively (Table 1).
DNA amplification: Result showed that the two samples were successfully amplified. The amplified products were found within 700-800 bp. corresponding to the ladder (Fig. 2).
Amplicons sequencing and sequence analysis: The MatK gene amplicons of the two samples were successfully sequenced. The result showed that samples A and B had sequence length of 755 and 766 bp, respectively (Fig. 3). Further, the result revealed the presence of 728 conserved codons and 38 single nucleotide polymorphisms (SNP) between sample A and sample B. The SNPs comprises of 20 point mutations, 11 to indels mutations and seven (7) un-sequenced codons. Table 2 showed the SNP between samples A and B.
Similarly, 251 and 255 amino acids residues were obtained for samples A and B respectively. These include 233 conserved residues and 22 mutations. The mutations comprises of seven (7) indels mutations, 13 point mutations and 2 untranslated codons. Table 3 showed points of mutation in the amino acids sequences of samples A and B.
Sequence identification: The BLAST results showed that the MatK sequence of sample A exhibited the highest similarity (99.2%) with T. tetraptera while sample B exhibited the highest similarity (99.71%) in congruence manner, with sequences of A. adiantifolia and A. petersiana congruently (Table 4).
From the phylogenetic tree, it was observed that sample A nested with T. tetraptera while sample B nested closely with A. adiantifolia in the same branch that contains A. petersiana (Fig. 4).
The genetic distance between sample A and Tetraptera sp. in the phenogram was between 0.1-0.4 and 0.1-0.6% between sample B and Albizia adiantifolia. The intraspecific distances were 0.1-0.3% for T. tetraptera, 0.40% for A. adianthifolia and 0.00% for A. petersiana. Table 5 shows the result.
The phylogenetic tree was constructed using Maximum Likelihood method based on the Tamura-Nei mode. The number above branches corresponds to bootstrap support.
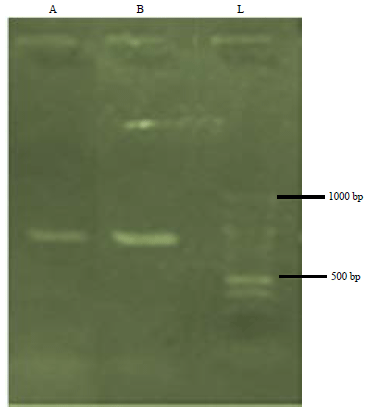 | |
| Fig. 2: | Agarose gel electrophoresis showing the amplified MatK gene of samples A and B |
L: 1 kb DNA/molecular ladder | |
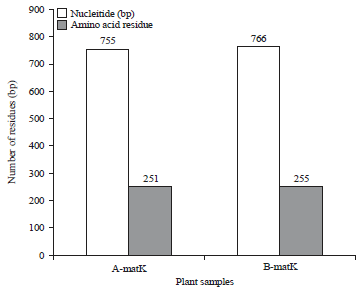 | |
| Fig. 3: | Characterization of nucleotide and protein sequence of samples A and B |
| Table 1: | DNA extraction and quantification results |
 | |
A260: DNA absorbance at A260 nm wavelength, A280 (nm): Protein absorbance at A280 nm wavelength, 260/280: Nucleic acid purity | |
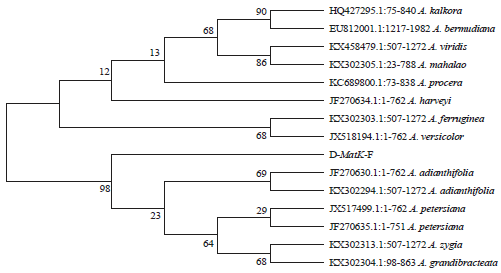 | |
| Fig. 4: | Phylogenetic tree for the identification of samples A and B |
| Table 2: | Single nucleotide polymorphisms (SNP) in MatK gene of samples A and B |
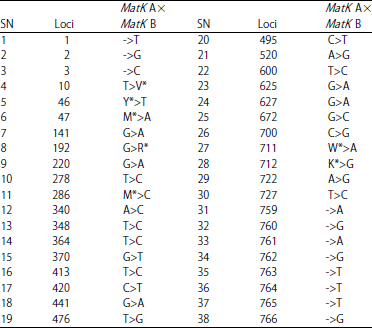 | |
-: Indels (insertion/deletion), Mismatch: Point mutations, *: Un-sequenced coding region, T: Thymine, G: Guanine, C: Cytosine, A: Adenine, M: Methionine, Y: Tyrosine, V: Valine, R: Arginine, K: Lysine, W: Tryptophan | |
| Table 3: | Mutations in MatK gene amino acid sequence of samples A and B |
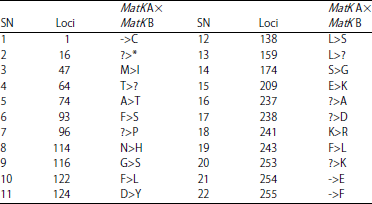 | |
*, ?: Non-transcribed region, -: Indels (insertion/deletion), Mismatches:Point mutations, T: Threonine, G: Glycine, C: Cysteine, A: Adenine, M: Methionine, Y: Tyrosine, V: Valine, R: Arginine, K: Lysine, W: Tryptophan | |
DISCUSSION
This study assessed the applicability of DNA barcoding (using MatK gene) for discrimination of two morphologically similar Mimosoideae species. The result showed that the total DNAs of sample A and B were below purity limit of 1.8, suggesting the presence of protein contaminants in the DNA isolate22. The weak purity may be due to the presence of aromatic rings in the purine and pyrimidine23. This observation faulted the opinion of chemical defenses such as tannins and phenols as the common contaminants of plant DNA24,25.
The amplicons of samples T. tetraptera and Albizia adianthifolia were found in the region of 750-800 base pairs corresponding to the ladder (Fig. 2). This is similar to Udensi et al.24 who reported 750 and 607 bp, respectively with the MatK region. This implies that the MatK gene of the studied samples were suitable for sequencing. Similar results using MatK was also observed24,26.
The obtained sequence of Sample A and B had 755 and 766 bp, respectively and 251 and 255 amino acid residues respectively. Moreover, sequence length that varied between 508 and 867 bp with an average of 803 bp and maintained that 500 bp is acceptable for the submission to BOLD database23. Also, according to Nithaniyal et al.25, sequence length of 300 bp satisfied the criterion to facilitate amplification. Therefore, the MatK gene of the samples adequately satisfied the requirement to be used as barcode region.
Results also revealed 11 bp for Sample A and B (4 for A. adianthifolia) residue differences in nucleotide and amino acid sequences. Most genetic variation is considered neutral but single base changes in and around a gene can affect its expression or the function of its protein products.
| Table 4: | BLAST authentication of samples A and B |
| Table 5: | Evolutionary distance of samples A and B in relation to Albizia species and Tetrapleura tetraptera based on MatK gene |
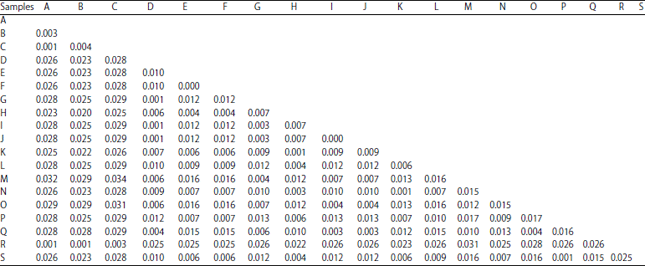 | |
A: AF521864.1: T. tetraptera, B: AF521865.1: T. tetraptera, C: MatK-F, D: MatK-F, E: EU812001.1: A. bermudiana, F: HQ427295.1: A. kalkora, G: JF270630.1: A. adianthifolia, H: JF270634.1: A. harveyi, I: JF270635.1: A. petersiana, J: JX517499.1: A. petersiana, K: JX518194.1: A. versicolor, L: KC689800.1: A. procera, M: KX302294.1: A. adianthifolia, N: KX302303.1: A. ferruginea, O: KX302304.1: A. grandibracteata, P: KX302305.1: A. mahalao, Q: KX302313.1: A. zygia, R: KX302355.1: T. tetraptera, S: KX458479.1: A. viridis | |
A single base change in coding region of nucleotide can result in amino acid change in the corresponding protein, hence if a point mutation alters protein function, the change can have drastic phenotypic and evolutionary consequences and beneficial mutations can sweep through the population and become fixed, thus contributing to speciation25. Also equally attributed variations in sequence lengths among taxa to indels mutations have accumulated effect during evolution27.
The extent of Indel mutations recorded implied that both samples evolved at different periods, while the 91.4% amino acid sequence similarity is suggestive of common ancestry (Table 4). It is plausible that both species though evolved at different times are closely related. Thus, organisms with high percentage sequence similarity in their genes have a similar pattern of evolution15,27. Sequence similarity implies both sequences shared common evolutionary ancestor. If two sequences have sequence identity greater than 70%, the implication is that they have about 90% probability or more to share the same biological processes and functions27. It is expected that the protein in both samples should have similar functionality since they retain high percentage identity in their nucleotide and amino acid sequence27,28. It thus suggests that MatK gene in both samples share very similar structural features owing to the high percentage similarity in their amino acid sequences despite their generic differences.
Samples A and B were identified to species and genus level respectively during the BLAST. Similar rates of sequence recoverage with MatK have been reported by Udensi et al.24, Cole et al.28 and Figueira et al.29 that no single gene locus has high levels of universality and resolvability, therefore, that proposed the use of multiple loci to increase success. Sample A was unambiguously identified as T. tetraptera, implying that the MatK adequately discriminated the species from its homologues. Figueira et al.29 attributed the discriminating power of any barcode region to availability of barcode gaps between the sequences. The existence of barcoding gap provides assurance that the genetic distance can be used to determine nomenclatural identity of an unknown specimen. For example, by observing taxon closes to the specimen in a phylogenetic tree27-29. Observations recorded similar successes with MatK gene in Fabaceae family. While different studies reported by Osman et al.20, Kamal and Klein21, Tallei et al.22, Udensi et al.24 and Thompson et al.30 the success among different angiosperm clades.
On the other hand, MatK of sample B had congruent identity with A. adiantifolia and A. petersiana. This indicates identification ambiguity, which may be due to insufficient barcode gaps. To resolve such ambiguity30, phylogenetic tree could be applied. The phylogenetic tree constructed showed that samples A (now identified as T. tetraptera) and B nested on separate branches of the tree indicating their different taxonomic affinities. However, the tree revealed that sample B is more closely related to A. adiantifolia than A. petersiana. The bootstrap values ranged between 44 and 100%, indicating a strong evolutionary relationship among Albizia and Tetrapleura taxa and a reliable identification model30.
Similarly, result showed that the genetic distance between sample A and B was 2.3%. This percentage variation is within the threshold for placement of taxa within generic rank. However, the genetic distance shown in the NCBI database between sample A and T. tetraptera ranged between 0.1-0.4% while that between sample B and A. adiantifolia was 0.1-0.6%. These satisfied the requirement to placing organisms in the same specific epithet30. On the other hand, the intraspecific distances were 0.1-0.3% for T. tetraptera, 0.40% for A. adianthifolia and 0.00% for A. petersiana. This satisfied the minimum requirements for species retention using a single DNA marker28,30. These results indicate highly reliable taxonomic identities for samples A and B.
Furthermore, the inter-genera distance between Tetraptera and Albizia ranged between 2.0 and 3.2% while the inter-specific distance ranged between 0.00 and 1.70% for Albizia genus. Plants adapt to harsh climatic conditions and variety of anthropological activities that may affect their survival by developing different survival characteristics and molecular diversity30. These molecular diversities are exhibited in the DNA barcodes as slight variations among individuals of same species. For instance, that shown in the NCBI database between two accessions (Table 5) of A. adianthifolia was 0.4% while that for T. tetraptera was 0.3%. These slight variations are products of specific environmental conditions and hence are used in identifying origins of taxa. It must be noted however that DNA barcoding operates on the assumption that selected gene region possess reduced variation within-taxon than between-taxa (www.barcodinglife. org) and the technique is potent in resolving slightest variations among taxa31,32.
The study further established the two specimens as separate taxa while morphometric analysis could then be applied to the specimens for rapid field authentication process. The study also affirmed that taxa exhibiting less than 97% homology cannot be grouped as same species. It is expected that other morphologically similar taxa be subjected to DNA analyses for accurate authentication.
CONCLUSION
From this study we conclude that two morphologically similar samples were successfully identified as T. tetraptera and A. adianthifolia using sequence BLAST and phylogenetic distance analyses respectively. Same markers recommended for use in the identification of other morphologically similar taxa.
SIGNIFICANCE STATEMENT
This study resolved the nomenclatural ambiguities between Tetraptera Tetrapleura and Albizia adianthifolia stemming from their morphological similarities. This study will therefore aid applications of phytochemical protocols to rapidly and cheaply but efficiently discriminate between the two species during field exercises. This will help users and researchers situate the accurate moieties contained in each species to enhance pharmacological and industrial endeavors. It will also jump start the phasing out process of expert recognition method in voucher specimen practice with one authenticated by DNA bar coding.
ACKNOWLEDGMENT
The authors appreciate the efforts of Prof Tafteng and staff of Mifor Consult, Calabar.
REFERENCES
- Ebigwai, J.K., M.T. Akesa and E. Ebigwai, 2019. Application of phytochemical protocols in authenticating six morphologically identical mimoisoidea members. Int. J. Scient. Eng. Res., 10: 609-621.
Direct Link - Ebigwai, J.K. and O.C. Enudi, 2019. Taxonomic authentication of two morphologically identical Senna species using Matk DNA barcoding and phytochemical protocol. Int. J. Scient. Eng. Res., 10: 629-644.
Direct Link - Gonzalez, M.A., C. Baraloto, J. Engel, S.A. Mori and P. Petronelli et al., 2009. Identification of Amazonian trees with DNA barcodes. PLoS ONE, Vol. 4.
CrossRefDirect Link - Kurnik, D., M. Muszkat, G.G. Sofowora, E.A. Friedman and W.D. Dupont et al., 2008. Ethnic and genetic determinants of cardiovascular response to the selective α2-adrenoceptor agonist dexmedetomidine. Hypertension, 51: 406-411.
CrossRefDirect Link - Abdel-Latif, A. and G. Osman, 2017. Comparison of three genomic DNA extraction methods to obtain high DNA quality from maize. Plant Methods, Vol. 13.
CrossRefDirect Link - Feng, J., D. Jiang, H. Shang, M. Dong and G. Wang et al., 2013. Barcoding poplars (Populus L.) from Western China. PLoS ONE, Vol. 8.
CrossRefDirect Link - Ford, C.S., K.L. Ayres, N. Toomey, N. Haider and A.J. Kelly et al., 2009. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc., 159: 1-11.
CrossRefDirect Link - van Huan, H., H.M. Trang and N. van Toan, 2018. Identification of DNA barcode sequence and genetic relationship among some species of Magnolia family. Asian J. Plant Sci., 17: 56-64.
CrossRefDirect Link - Hollingsworth, P.M., 2008. DNA barcoding plants in biodiversity hot spots: Progress and outstanding questions. Heredity, 101: 1-2.
CrossRefDirect Link - Birdi, K., C. Clegg, M. Patterson, A. Robinson, C.B. Stride, T.D. Wall and S.J. Wood, 2008. The impact of human resource and operational management practices on company productivity: A longitudinal study. Personnel Psychol., 61: 467-501.
CrossRefDirect Link - Liu, K., A.A. Abdullah, M. Huang, T. Nishioka, M. Altaf-Ul-Amin and S. Kanaya, 2017. Novel approach to classify plants based on metabolite-content similarity. BioMed Res. Int., Vol. 2017.
CrossRefDirect Link - Kang, Y., Z. Deng, R. Zang and W. Long, 2017. DNA barcoding analysis and phylogenetic relationships of tree species in tropical cloud forests. Scient. Rep., Vol. 7, No. 1.
CrossRefDirect Link - Duret, L., J. Cohen, C. Jubin, P. Dessen and J.F. Gout et al., 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: A somatic view of the germline. Genome Res., 18: 585-596.
CrossRefDirect Link - Shinwari, Z.K., K. Jamil and N.B. Zahra, 2014. Molecular systematics of selected genera of subfamily Mimosoideae-Fabaceae. Pak. J. Bot., 46: 591-598.
Direct Link - Wattoo, J.I., M.Z. Saleem, M.S. Shahzad, A. Arif, A. Hameed and M.A. Saleem, 2016. DNA barcoding: Amplification and sequence analysis of rbcl and matK genome regions in three divergent plant species. Adv. Life Sci., 4: 3-7.
Direct Link - Mishra, P., A. Kumar, V. Rodrigues, A.K. Shukla and V. Sundaresan, 2016. Feasibility of nuclear ribosomal region ITS1 over ITS2 in barcoding taxonomically challenging genera of subtribe Cassiinae (Fabaceae). PeerJ, Vol. 4.
CrossRefDirect Link - Osman, M., F. Golam, S. Saberi, N.A. Majid, N.H. Nagoor and M. Zulqarnain, 2011. Morpho-agronomic analysis of three roselle (Hibiscus sabdariffa L.) mutants in tropical Malaysia. Aust. J. Crop Sci., 5: 1150-1156.
Direct Link - Kamal, M.A. and P. Klein, 2011. Determination of sugars in honey by liquid chromatography. Saudi J. Biol. Sci., 18: 17-21.
CrossRefDirect Link - Tallei, T.E., P.D. Irawan and B.J. Kolondam, 2016. DNA barcoding analysis of matK gene of some Syzygium species. Proceedings of the Bioinformatics Workshop 2016: Developing Knowledge and Skill in Bioinformatics for Young Indonesian Scientists in Improving Research Quality in Life Science and Sustainable Exploration of Biodiversity in Indonesia, September 13-15, 2016, Al Azhar University, Jakarta, Indonesia.
- Bell, K.L., N. de Vere, A. Keller, R.T. Richardson, A. Gous, K.S. Burgess and B.J. Brosi, 2016. Pollen DNA barcoding: Current applications and future prospects. Genome, 59: 629-640.
CrossRefDirect Link - Udensi, O.U., N.E. Edu, E.V. Ikpeme, O.O. Onung, L.I. Emeagi, B.I. Nwanze and E.R. Ejiyere, 2017. Genotyping of pigeon pea [Cajanus cajan (L.) Millsp.] accessions obtained from International Institute of Tropical Agriculture (IITA) germplasm using random amplified polymorphic DNA. J. Exp. Agric. Int., 17: 1-12.
CrossRefDirect Link - Nithaniyal, S., S.G. Newmaster, S. Ragupathy, D. Krishnamoorthy, S.L. Vassou and M. Parani, 2014. DNA barcode authentication of wood samples of threatened and commercial timber trees within the tropical dry evergreen forest of India. PLoS ONE, Vol. 9.
CrossRefDirect Link - Bhuiyan, N.H., G. Selvaraj, Y. Wei and J. King, 2009. Gene expression profiling and silencing reveal that monolignol biosynthesis plays a critical role in penetration defence in wheat against powdery mildew invasion. J. Exp. Bot., 60: 509-521.
CrossRefDirect Link - Sarvananda, L., S.S.A. Abed, J. Rohini and B. Sathyamurthy, 2016. Molecular identification of the medicinal plant Justicia gendarussa using matk gene. Eur. J. Pharmaceut. Med. Res., 3: 259-266.
Direct Link - Cole, T.B., J. Coburn, K. Dao, P. Roque and Y.C. Chang et al., 2016. Sex and genetic differences in the effects of acute diesel exhaust exposure on inflammation and oxidative stress in mouse brain. Toxicology, 374: 1-9.
CrossRefDirect Link - Figueira, V., I. Vaz-Moreira, M. Silva and C.M. Manaia, 2011. Diversity and antibiotic resistance of Aeromonas spp. in drinking and waste water treatment plants. Water Res., 45: 5599-5611.
CrossRefDirect Link - Thompson, W., C. Thakar, D.J. Rolton, J. Wilson-MacDonald and C. Nnadi, 2016. The use of magnetically-controlled growing rods to treat children with early-onset scoliosis: Early radiological results in 19 children. Bone Joint J., 98: 1240-1247.
CrossRefDirect Link - Wei, Y.N., X.M. Wang, P.C. Yao, X.Y. Chen and H.Q. Li, 2017. Comparison of species resolution rates of DNA barcoding for Chinese coastal halo-tolerant plants. Biodivers. Sci., 25: 1095-1104.
CrossRefDirect Link - Fazekas, A.J., K.S. Burgess, P.R. Kesanakurti, S.W. Graham and S.G. Newmaster et al., 2008. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE, Vol. 3.
CrossRefDirect Link