
C.P. Kumar
Centre for Advanced Study in Marine Biology, Faculty of Marine Biology, Annamalai University, Parangipettai, Tamil Nadu, India
B.A. John
Department of Biotechnology, Kulliyyah of Science, International Islamic University Malaysia, Jalan Sultan Ahmad Shah, Bandar Indera Mahkota, 25200, Kuantan Pahang, Malaysia
S.A. Khan
Centre for Advanced Study in Marine Biology, Faculty of Marine Biology, Annamalai University, Parangipettai, Tamil Nadu, India
P.S. Lyla
Centre for Advanced Study in Marine Biology, Faculty of Marine Biology, Annamalai University, Parangipettai, Tamil Nadu, India
A.S. Kamaleson
Division of Biological Oceanography, National Institute of Oceanography, Dona Paula, Goa, India
International Journal of Virology
Year: 2011 | Volume: 7 | Issue: 2 | Page No.: 42-52







ABSTRACT
Recent emergence of Chikungunya virus has attracted the researchers to explore its whole genome to predict the structural changes at the genetic level during disease outbreak. Present study was aimed to investigate the mutational patterns at the genome level in Chikungunya virus using advanced software programs. Pair-wise analysis of whole genome (n = 84) of Chikungunya virus revealed that structural genes mutated 2.6 times higher than non-structural genes. The chronological analysis showed that the Chikungunya virus prevalent during 2008 mutated higher than any other outbreaks recorded so far. Number of singleton sites in the variable region of viral genome was found higher when compared to number of parsimonous sites. Chronological and geographical scale analysis of the viral outbreak showed that about 30% of viral genome was subjected to mutation so far. Chikungunya virus isolated from Malaysian geography had underwent more mutations when compared anyother geographical locations. Highest number of singleton mutations were observed in Chikungunya viruses isolated from Malayasian geography whereas highest number of parsimonous informative sites were observed in Chikungunya isolates of India. The mutational analysis also revealed that viral genome contained two times more singleton sites compared to parsimonious sites. About 1207 nucleotides in the structural protein coding region of Chikungunya virus genome was identified as a unique DNA signature witch could be used in precise and fast diagnosing of Chikungunya virus in future.
PDF Abstract XML References Citation
Received: May 20, 2011;
Accepted: June 08, 2011;
Published: August 01, 2011
How to cite this article
C.P. Kumar, B.A. John, S.A. Khan, P.S. Lyla and A.S. Kamaleson, 2011. Mutational Patterns and DNA Barcode for Diagnosing Chikungunya Virus. International Journal of Virology, 7: 42-52.
DOI: 10.3923/ijv.2011.42.52
URL: https://scialert.net/abstract/?doi=ijv.2011.42.52
DOI: 10.3923/ijv.2011.42.52
URL: https://scialert.net/abstract/?doi=ijv.2011.42.52
INTRODUCTION
Chikungunya virus (CHIKV) is a mosquito-transmitted Alphavirus belonging to family Togaviridae (Schuffenecker et al., 2006; Cavrini et al., 2009; Chakkaravarthy et al., 2011). The genus Alphavirus consists of 29 distinct species. Like all Alphaviruses, CHIKV has a genome consisting of a linear, positive-sense, single-stranded RNA molecule of approximately 11.8 kb (Vanlandingham et al., 2005). The scope and magnitude of the 2005-2007 CHIKV outbreaks has led to speculation that the virus has been mutated to a more virulent form (Greene et al., 2005).
Previous studies have confirmed that when an outbreak occurs in a given region, the sequence of the virus associated with the epidemic is genetically aligned with other strains and thus forms identical isolations (Powers and Logue, 2007). Phylogenetic studies of CHIKV revealed that two distinct CHIKV lineages were delineated; one containing all of the available isolates from West Africa and the second comprising all South and East African strains as well as isolates from Asia. Within this second lineage, Asian strains got grouped together in a genotype distinct from the African groups. Additionally, paraphyletic grouping of the African sequences in phylogenetic trees corroborated historical evidence that CHIKV originated in Africa and subsequently was introduced into Asia (Powers et al., 2000). In Malaysia, the first outbreak was reported in the year 1998, later on it became epidemic and affected 51 people (Ali et al., 2011).
DNA barcoding is a molecular taxonomic method which uses a short genetic marker in an organism’s genome to identify it as a particular species (Hebert et al., 2004). DNA barcoding is the term used to describe a method proposed for producing a unique identifier for all living species (Hebert et al., 2003). Identifying species through DNA barcoding is also helpful for understanding interspecies interactions and to delineate morphologically cryptic taxa (Pfenninger et al., 2007; Tedersoo et al., 2007; Kamaruzzaman et al., 2011; Khan et al., 2010; Kumar et al., 2011). The resulting barcode sequences can be used for both species identification and for limited phylogenetic analysis to aid in placing those organism not already represented in the database. This approach has already led to number of publication representing studies of a variety of animal groups (Md-Zain et al., 2010). Empirical support for the barcoding concept ranges from studies of invertebrates (e.g. springtails and butterflies) to birds (Hogg and Hebert, 2004).
Previously single gene approach (structural E1 gene) has been largely used to study molecular characterization and mutational patterns of Chikungunya virus. Furthermore, datasets that include smaller fragments of the E1 gene (~300 nt) are significantly less robust (Powers and Logue, 2007). This diagnosis is mainly based on the amplification of E1 gene by PCR reaction (Pfeffer et al., 2002). PCR systems are not always reliable as pseudo-amplifications are of frequent occurrence. Hence sequencing the signature gene portions (DNA barcodes) would greatly facilitate the rapid and accurate identification. The analysis of whole genome of Chikungunya virus was found promising and would be necessary to derive pivotal conclusion regarding the mutational patterns and DNA barcode of Chikungunya virus (Santhosh et al., 2008).
Utilization of DNA barcodes in diagnostic protocols are of recent interest. Defining and producing such DNA barcodes to diagnose pathogens would vitally improve standards of diagnosing practices. DNA barcodes for identifying virus especially human virus should be defined because when bought into practice the technique will greatly improvise human health and prevention of diseases (Shankar et al., 2009). Taking this problem into consideration the present study employs analyzing the whole genome of Chikungunya virus along with its sister taxa to define DNA barcodes for Chikungunya virus.
MATERIALS AND METHODS
Viral genome retrieval: Eighty four whole genomes available (until September 2010) were retrived from NCBI (National Centre for Biotechnolical Information) and taken for analysis. Information of sequence accession numbers, its geography and year of sequence are tabulated (Table 1). The viral genomic analysis was carried out by grouping entire viral genes in to two groups, one containing all structural genes and other containing non-structural genes of the virus.
| Table 1: | List of Chikungunya genome retrieved from Genbank and its accession number, geography and time of publication were tabulated |
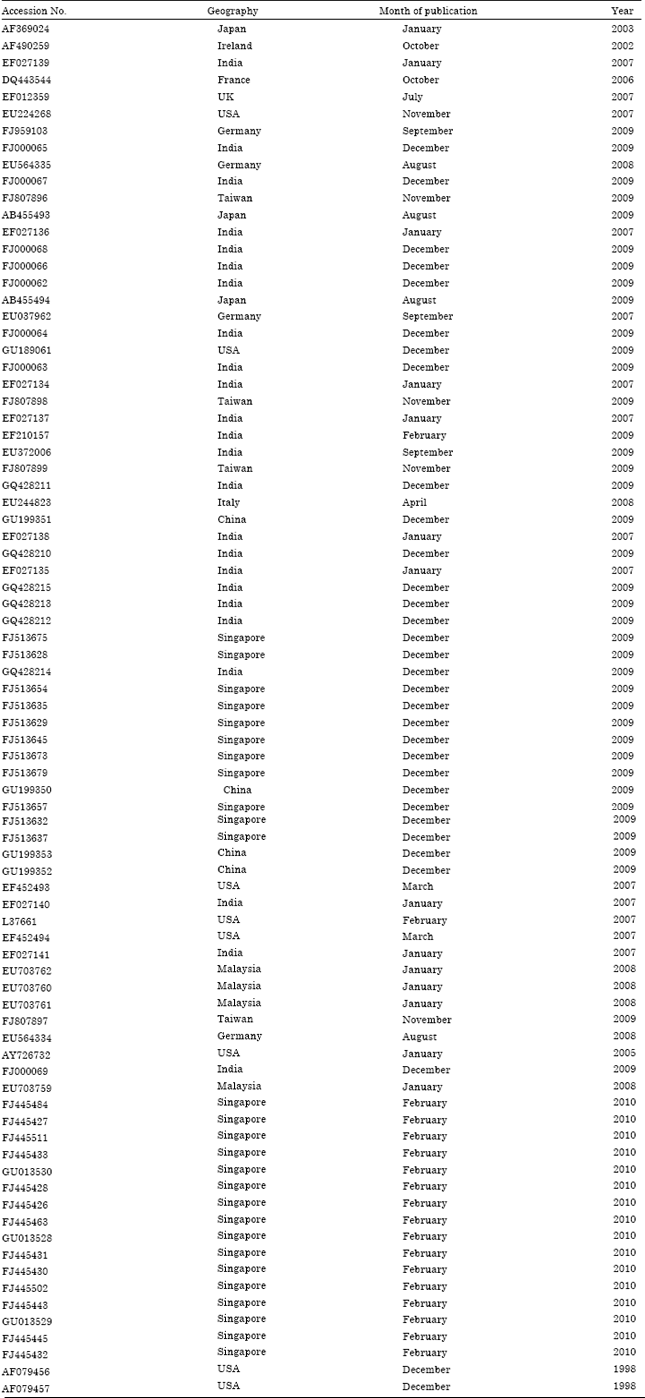 | |
Mutational analysis: The viral genomes (n = 84) were aligned in MEGA (Molecular Evolutionary Genetic Analysis) ver. 4. 0 (Tamura et al., 2007). The pair-wise distance data of structural genes and non-structural genes was calculated using Kimura-2 parameter distance (Nei and Kumar, 2000). The aligned whole genome was carefully scanned for conserved sites (a site containing the same nucleotide in all sequences analyzed through alignment), variable sites (contains at least two types of nucleotides all sequences analyzed through alignment). Variable sites can be singleton or parsimony-informative. singleton sites (a singleton site contains at least two types of nucleotides with, at most, one occurring multiple times) and parsimoniously informative sites (a site is parsimony-informative if it contains at least two types of nucleotides and at least two of them occur with a minimum frequency of two) in viral genome across chronological (from 2002 to 2010) and geographical scales (from sequences of 13 different countries) were calculated (Goldman, 1993).
Defining DNA signatures for chickungunya virus: CHIKV genome was aligned with its closest relatives; O’nyong-nyong virus and Igbo Ora virus (Khan et al., 2002) of genus Alpha virus using Clustal ver. 2.0 (Larkin et al., 2007). The aligned regions were carefully scanned to detect variable sites specific to Chikungunya virus.
RESULTS AND DISCUSSION
Pair-wise distance within structural, non-structural and whole genome: The genome of the virus was analysed under two heads, structural genes and non-structural genes. It was found that the structural genes of Chikungunya virus mutate more (0.057) rapidly compared to non-structural genes (0.022) (Fig. 1). Through pair-wise analysis using Kimura2-parameter, it was found that structural genes undergoes 2.6 times more mutation than non-structural genes of viral genome. This structural gene variations was well known to play vital role in shuffling viral antigenic properties which qualifies the virus to resist host’s immune system.
Mutational patterns across chronological and geographical scales: The genomes of Chikungunya virus was chronologically and geographically grouped to study its conserved and variability patterns in the viral genome. It was assumed that the publication date of the viral genome in National Center for Biotechnology Information (NCBI) coressponds to geneome generated within one year of Chikungunya disease in the specified geography. The mutatioanal analysis was carried out by determining number of conserved site and variable sites in the viral genome. The variable sites (mutating sites) contained both the types of variation i.e., singleton sites and parsimonous informative sites.
The chronological analysis showed that the out-break of Chikungunya virus in 2008 mutated higher than any other out-breaks (due to less conserved region [72.5%] and high variable region [26.4%]) recorded so far (Fig. 2). This observation was well corresponded with previous study where positive correlation was observed between higher mutational rate in 2008 and major out breaks of Chikungunya disease in 2007-2008 (Huang et al., 2009; Powers and Logue, 2007; Powers et al., 2000). Across the chronological and geographical scale of viral outbreak, number of singleton sites was found higher compared to number of parsimonous sites in the variable region of viral genome (Fig. 3a, b). Among the mutation sites analysed across geographical and chronological scales, viral genome contained two times more sigleton sites compared to parsimonious sites.
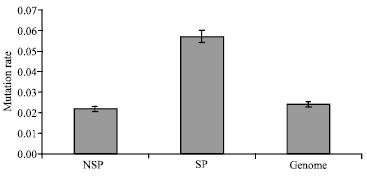 | |
| Fig. 1: | Genetic distances with the structural, non-structural and the whole genome of the Chickungunya virus. Over all; structural genes undergoes 2.6 times more mutation than the non-structural genes in Chickungunya virus. NSP: Non structural protein; SP: Structural protein |
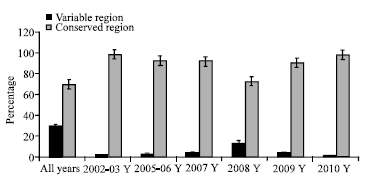 | |
| Fig. 2: | Percentage of conserved region and variable regions in the whole genome of Chikungunya virus across the time scale |
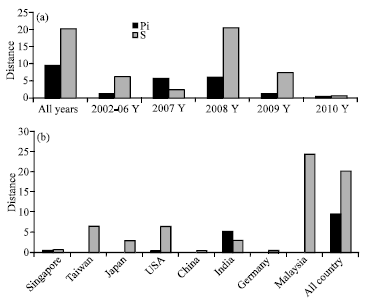 | |
| Fig. 3(a-b): | (a) Parsimonious and singleton distance in the variable regions of Chikungunya virus genome collected across the time scale and (b) Parsimonious distance and singleton distance in the variable regions of Chikungunya virus genome collected across the Geography. Pi: Parsimonious distance; S: Singleton distance |
Overall mutational analysis showed that about 30% of viral genome has undergone mutation so far (singleton sites mutation (20%) +parisimonious sites mutation (9.4%) = ~ 30%) (Fig. 4).
When analyzed geographically, it was found that Chikungunya virus outbreaks at Malaysian geography has accumulated more mutations (due to high variable region = 24.1%) compared to any other country (Fig. 5). Next to Malaysia, Indian outbreaks contained more mutations (variaable region = 7.3%). When this analysis was further resolved into parsimonous sites and singleton sites, it was found that Malayasian out-breaks contained more sigleton mutations whereas Indian out-breaks contained more parsimonous type of mutation. A research evidence of Chikungunya virus imported in Malaysia from India was also recorded (Lam et al., 2001; Noridah et al., 2007). An interesting question might be what sort of environmental factor in Malaysia influenced the change in mutational pattern of imported Indian strain, as the imported strain showed reduced number of parsimonious informative sites and increased number of singleton site in the viral genome.
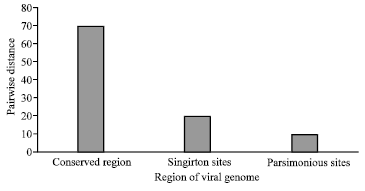 | |
| Fig. 4: | Bar diagram briefing the quantity of mutational region (variable region) and conserved regions in the viral genome. It was found that about 30% of viral genome underwent mutation so far which is further described as singleton sites and parisimonious sites |
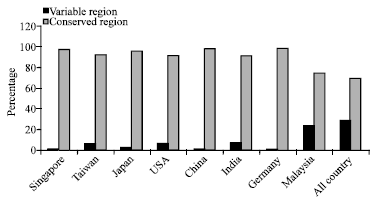 | |
| Fig. 5: | Percentage of variation and conservation in the whole genome of Chikungunya recorded cross the geography |
It is quite evident that the environmental factors influencing mutation in virus should be studied in more detailed fashion to develop preventive measures against Chikungunya virus.
Defining chikungunya viral DNA barcode: Multiple alignment was done using whole genome of all members of Togaviridae (18 species). The multiple alignment analysis showed that 408 nucleotides in the structural protein region of Chikungunya virus could be a potential barcode to detect the virus during outbreaks. The region contained about 20 nucleotide insertions unique to Chikungunya virus and was absent among its closest relative (Onyong nyong virus and Igbo ora virus) (Fig. 6). The defined 408 bps of DNA barcode was subjected to conserved domain BLAST search to detect the function of the gene (Geer et al., 2002; Marchler-Bauer et al., 2007; Marchler-Bauer, 2009). The defined DNa barcode code for S3-peptidase (structural polyprotein).
Chikungunya virus has been responsible for human morbidity for several hundred years. CHIKV genome could be segregated in to two major portions, viz Structural (3746 bps) genes and Non-structural genes (7502 bps). All available 84 CHIKV genomes (until June 2010) retrieved from NCBI was used in the study. The Pair-wise distance due to mutational events in structural protein coding genes (SP), Non-Structural (NSP) protein coding genes and the whole genome of the members of CHIKV was calculated. Over all, structural genes underwent 2.6 times more mutation than the non-structural genes in CHIKV genome prevalent during the outbreaks between 2003 and 2010.
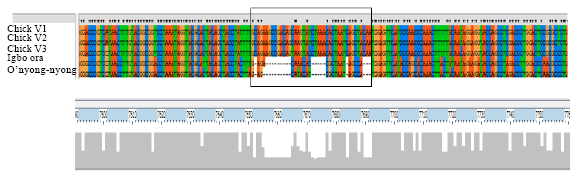 | |
| Fig. 6: | Multiple alignment of whole genome of Chikungunya virus along with its sister taxa aligned using Clustal X. The square box in the alignment indicates the presence of insertion sequences Chikungunya viral genome. The absence of such inserts among sister taxa of Chikungunya virus was indicated by gaps (bottom two sequences). This specified signature in the capsid gene of viral genome was proposed as DNA barcode for Chikungunya virus. |
The genomes of CHIKV was chronologically and geographically grouped to study their conserved and variability patterns in the viral genome. It was found that in 2008 infection, CHIKV underwent highest mutational rate compared to any other outbreaks of Chikungunya virus recorded so far. Interestingly positive correlation was observed beteween higher mutational rate in 2008 and major outbreak during 2007-2008. This may lead to a hypothesis that intensity of mutational rate is directly propotional to the intensity of outbreaks.
While analysing variable sites across geographical and chronological scale, it was found that number of singleton sites was higher than Parsimonous information sites in the viral genome. This fact was also found true during analysis of mutational pattern of Chikungunya genome prevalent during the outbreak in 2007-2008. When analyzed geographically, it was found that CHIKV outbreaks at Malaysian geography has accumulated more mutations compared to any other outbreak in other countries put together. A notable fact was that next to Malaysia, Indian out breaks contained more mutations. When parsimonous sites and singleton informative sites in the variable region were geographically analyzed, Malayasian out-breaks contained maximum sigleton mutations, whereas Indian out-breaks contained maximum parsimonous type of mutation. It is imperative that the environmental factors influencing mutation of virus should be focused more in future to develop preventive measures against Chikungunya virus.
Togaviridae genome (18 members) was aligned (data not shown) using Clustal X for DNA signature analysis. The process was found complicated due to the presence of randomly placed indels across the viral genome. Hence only the closest relative of Chikungunya virus (Igbo ora virus and Onyong nyong virus) was considered for alignment analysis. It was found that 1207 nucleotides in the structural protein region of Chikungunya virus could be a potential barcode as it was found unique in CHIKV genome. The region contained about 20 nucleotide insertion unique to CHIKV which was absent in to closest relatives of CHIKV viz., Onyong nyong virus and Igbo ora virus. Though only 20 nucleotides insert was found, 1207 nucleotides on the whole (including the regions flanking the signature sequences) were considered as DNA barcode. The region contained notable comparatively variations in Chikungunya genome other than the 20 nucleotide inserts. Also the conserved region flanking the variable region allows to develope primers for amplification and sequencing reaction. Also the proposed barcode if tested successfully in wet lab, will facilitate the delineation of Chikungunya virus from other members of Togaviridae during mixed outbreak crisis.
The 408 bps defined as DNA barcode is located in structural genes (which contained higher mutational rate) of viral genome. The region codes for S3-peptidase- a structural polyproteins (Cheng et al., 1995). However, this region is proposed only as a hypothetical barcode. Further wet lab testing has to be carried out to confirm the proposed phenomenon. The hypothetical DNA barcode discovered through this study was deposited in NCBI as secondary data and could be accessed through accession number HQ199837.
CONCLUSION
Over all, structural genes underwent 2.6 times more mutation than the non-structural genes in CHIKV genome. CHIKV underwent highest mutational rate in 2008 than any other outbreaks of Chikungunya virus recorded so far. Interestingly major outbreak of Chikungunya virus has occurred during 2007-2008. This may lead to a hypothesis that intensity of mutational rate is directly propotional to the intensity of outbreaks. However, experimental data was required to prove the phenonmenon. CHIKV outbreaks at Malaysian geography has accumulated more mutations compared to anyother geographical locations. Malayasian out-breaks contained maximum sigleton mutations, whereas Indian out-breaks contained maximum parsimonous type of mutation. It is imperative that the environmental factors influencing mutation of virus should be the focused more in future to develop preventive measures against Chikungunya virus. The hypothetical DNA barcode discovered through this study was deposited in NCBI and could be accessed through its number HQ199837. This barcode can play vital role in diagnosing CHIKV infection with accuracy and rapidity.
REFERENCES
- Cavrini, F., P. Gaibani, A.M. Pierro, G. Rossini, M.P. Landini and V. Sambri, 2009. Chikungunya: An emerging and spreading arthropod-borne viral disease. J. Infect. Dev. Ctries., 310: 744-752.
Direct Link - Chakkaravarthy, V.M., S. Vincent and T. Ambrose, 2011. Novel approach of geographic information systems on recent outbreaks of chikungunya in Tamil Nadu, India. J. Environ. Sci. Technol., 4: 387-394.
CrossRefDirect Link - Cheng, R.H., R.J. Kuhn, N.H. Olson, M.G. Rossmann, H.K. Choi, T.J. Smith and T.S. Baker, 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell, 80: 621-630.
PubMedDirect Link - Goldman, N., 1993. Statistical tests of models of DNA substitution. J. Mol. Evolut., 36: 182-198.
Direct Link - Greene, I.P., S. Paessler, L. Austgen, M. Anishchenko, A.C. Brault, R.A. Bowen and S.C. Weaver, 2005. Envelope glycoprotein mutations mediate equine amplification and virulence of epizootic venezuelan equine encephalitis virus. J. Virol., 79: 9128-9133.
Direct Link - Geer, L.Y., M. Domrachev, D.J. Lipman and S.H. Bryant, 2002. CDART: Protein homology by domain architecture. Genome Res., 12: 1619-1623.
PubMed - Hebert, P.D.N., M.Y. Stoeckle, T.S. Zemlak and C.M. Francis, 2004. Identification of birds through DNA barcodes. PLoS Biol., Vol. 2, No. 10.
CrossRefDirect Link - Hebert, P.D.N., S. Ratnasingham and J.R. Dewaard, 2003. Barcoding animal life: Cytochrome c oxidase divergences among closely related species. Proc. Roy. Soc. Lond. Biol. Sci., 270: 596-599.
PubMedDirect Link - Hogg, I.D. and P.D.N. Hebert, 2004. Biological identification of springtails (Hexapoda: Collembola) from the Canadian Arctic, using mitochondrial DNA barcodes. Can. J. Zool., 82: 749-754.
CrossRefDirect Link - Huang, J.H., C.F. Yang, C.L. Su, S.F. Chang and C.H. Cheng et al., 2009. Imported chikungunya virus strains, Taiwan, 2006-2009. Emerg. Infect. Dis., 15: 1854-1856.
PubMed - Kamaruzzaman, B.Y., B.A. John, K. Zaleha and K.C.A. Jalal, 2011. Molecular phylogeny of horseshoe crab. Asian J. Biotechnol., 3: 302-309.
CrossRefDirect Link - Khan, A.H., K. Morita, M.M.C. Parquet, F. Hasebe, E.G. Mathenge and A. Igarashi, 2002. Complete nucleotide sequence of chikungunya virus and evidence for an internal polyadenylation site. J. General Virol., 83: 3075-3084.
PubMed - Khan, S.A., P.S. Lyla, B.A. John, C.P. Kuamr, S. Murugan and K.C.A. Jalal, 2010. DNA barcoding of Stolephorus indicus, Stolephorus commersonnii and Terapon jarbua of Parangipettai coastal waters. Biotechnology, 9: 373-377.
CrossRefDirect Link - Lam, S.K., K.B. Chua, P.S. Hooi, M.A. Rahimah and S. Kumari, 2001. Chikungunya infection-an emerging disease in Malaysia. Southeast Asian J. Trop. Med. Public Health, 32: 447-451.
PubMedDirect Link - Larkin, M.A., G. Blackshields, N.P. Brown, R. Chenna and P.A. McGettigan et al., 2007. Clustal W and clustal X version 2.0. Bioinformatics, 23: 2947-2948.
CrossRefPubMedDirect Link - Marchler-Bauer, A., 2009. CDD: Specific functional annotation with the conserved domain database. Nucl. Acids Res., 37: 205-210.
CrossRefDirect Link - Marchler-Bauer, A., J.B. Anderson, M.K. Derbyshire, C. de Weese-Scott and N.R. Gonzales et al., 2007. CDD: A conserved domain database for interactive domain family analysis. Nucl. Acids Res., 35: D237-D240.
CrossRefDirect Link - Md-Zain, B.M., S.J. Lee, M. Lakim, A. Ampeng and M.C. Mahani, 2010. Phylogenetic position of Tarsius bancanus based on partial cytochrome b DNA sequences. J. Biol. Sci., 10: 348-354.
CrossRefDirect Link - Nei, M. and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, United Kingdom, ISBN-13: 9780195350517, Pages: 352.
Direct Link - Noridah, O., V. Paranthaman, S.K. Nayar, M. Masliza, K. Ranjit and I. Norizah, 2007. Outbreak of chikungunya due to virus of Central/East African genotype in Malaysia. Med. J. Malaysia, 62: 323-328.
Direct Link - Pfeffer, M., B. Linssen, M.D. Parker and R.M. Kinney, 2002. Specific detection of Chikungunya virus using a RT-PCR/Nested PCR combination. J. Vet. Med. B., 49: 49-54.
Direct Link - Pfenninger, M., C. Nowak, C. Kley, D. Steinke and B. Streit, 2007. Utility of DNA taxonomy and barcoding for the inference of larval community structure in morphologically cryptic Chironomus (Diptera) species. Mol. Ecol., 16: 1957-1968.
CrossRef - Powers, A.M., A.C. Brault, R.B. Tesh and S.C. Weaver, 2000. Re-emergence of chikungunya and O'nyong-nyong viruses: Evidence for distinct geographical lineages and distant evolutionary relationships. J. Gen. Virol., 81: 471-479.
PubMedDirect Link - Kumar, C.P., B.A. John, S.A. Khan, P.S. Lyla, S. Murugan, M. Rozihan and K.C.A. Jalal, 2011. Efficiency of universal barcode gene (Coxi) on morphologically cryptic mugilidae fishes delineation. Trends Applied Sci. Res., 6: 1028-1036.
CrossRefDirect Link - Santhosh, S.R., P.K. Dash, M.M. Parida, M. Khan, M. Tiwari and P.V. Lakshmana Rao, 2008. Comparative full genome analysis revealed E1: A226V shift in 2007 Indian Chikungunya virus isolates. Virus Res., 135: 36-41.
CrossRef - Schuffenecker, I., I. Iteman, A. Michault, S. Murri and L. Frangeul et al., 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLOS Med., 3: 263-263.
PubMed - Shankar, B.P., R.N.S. Gowda, B. Pattnaik, B.H.M. Prabhu, S.S. Patil and H.K. Pradhan, 2009. Rapid detection of highly pathogenic avian influenza H5N1 Virus by TaqMan reverse transcriptase-polymerase chain reaction. Int. J. Poult. Sci., 8: 260-263.
CrossRefDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Tedersoo, L., T. Suvi, K. Beaver and U. Koljalg, 2007. Ectomycorrhizae fungi of the seycheelles: Diversity patterns and host from the native Vateriopsis seychellarum (Dipterocarpaceae) and Intsia bijuga (caesalpiniaceae) to the introduced Eucalyptus robusta (Myrtaceae) but not Pinus caribea (Pinaceae). New Phytol., 175: 321-333.
Direct Link - Vanlandingham, D.L., K. Tsetsarkin, C. Hong, K. Klingler, K.L. McElroy, M.J. Lehane and S. Higgs, 2005. Development and characterization of a double subgenomic chikungunya virus infectious clone to express heterologous genes in Aedes aegypti mosquitoes. Insect. Biochem. Mol. Biol., 35: 1162-1170.
CrossRef