
Pankaj Bhatia
Swami Vivekanand College of Pharmacy, Banur, Patiala, 140401, India
Sushma Gupta
Swami Vivekanand College of Pharmacy, Banur, Patiala, 140401, India
Saurabh Sharma
ISF College of Pharmacy, Moga, India
International Journal of Pharmacology
Year: 2014 | Volume: 10 | Issue: 4 | Page No.: 200-212







ABSTRACT
Homocysteine (Hcy) excess as a result of impaired metabolism due to deficiency in cofactors (vitamin B6, B12, folate) or genetic alteration in metabolic enzymes (Methionine synthase, methyltetrahydrofolate reductase, cystathionine β-synthase and cystathionine-γ-lyase) lead to acquired metabolic anomaly known as hyperhomocysteinemia. Hyperhomocysteinemia (HHcy) is an independent major casual determinant of cerebrovascular and cardiovascular disorders viz., carotid artery disease, atherosclerosis, stroke and vascular dementia. The intense dysfunction of vascular endothelium following hyperhomocysteinemia has been implicated as a factor in the extension of pathological conditions. One of the therapeutic goals of modern vascular biology is to design strategies to limit vascular endothelium dysfunction. A sound understanding of the abnormality in homocysteine metabolic pathway and vascular endothelium dysfunction is necessary before a specific intervention is pursued. Summarized is the description of the homocysteine, its fate and abnormalities that leads to hyperhomocysteinemia. Further, the pathobiology of homocysteine excess which are noted to cause oxidative stress, impairment of intracellular transduction pathways, release of inflammatory mediators, dysfunction of endothelial progenitor cells and RBCs hemolysis is also discussed. Collectively these pathobiological mechanisms causes the decrease in cell survival, activation of proinflammatory mediators, apoptosis and consequently leads to vascular endothelium dysfunction. On the basis of the multiple toxic effects of homocysteine excess, interventions designed for these mechanisms may provide novel targets for the development of vascular protective agents.
PDF Abstract XML References Citation
Received: February 28, 2014;
Accepted: June 03, 2014;
Published: July 16, 2014
How to cite this article
Pankaj Bhatia, Sushma Gupta and Saurabh Sharma, 2014. Homocysteine Excess and Vascular Endothelium Dysfunction: Delineating the Pathobiological Mechanisms. International Journal of Pharmacology, 10: 200-212.
DOI: 10.3923/ijp.2014.200.212
URL: https://scialert.net/abstract/?doi=ijp.2014.200.212
DOI: 10.3923/ijp.2014.200.212
URL: https://scialert.net/abstract/?doi=ijp.2014.200.212
INTRODUCTION
Homocysteine is a naturally occurring nonproteogenic sulfur-containing amino acid produced as part of the body’s methylation process. The Hcy accumulated either in inherited disorders, that alters enzyme activity in metabolic transsulfuration and remethylation pathways or alternatively, in nutritional deficiencies of essential cofactors or enzyme substrates, including cobalamine (vitamin B12), folate or pyridoxine (vitamin B6), thus results in blockage of homocysteine metabolic pathways (Ingenbleek, 2011). Low level of homocysteine (Hcy) (5-14 μmol L-1) is normally found in the plasma (Sharifi et al., 2010). Homocysteine levels can range from a low level of 14 μmol L-1 to a devastating level of 100 μmol L-1 in severe cases (Seshadri et al., 2002). Hyperhomocysteinemia (HHcy) is an acquired metabolic anomaly which was first identified by McCully (Ingenbleek, 2011). The HHcy may leads to cardiovascular and cerebrovascular complications like atherosclerosis, thrombosis, stroke, impairs angiogenesis, Alzheimer’s disease, dementia, declining memory, poor concentration and judgment and lowered mood etc. Increase in homocysteine concentration is also crucially involved in various autoimmune and malignant diseases (Dalton et al., 1997). Hyperhomocysteinemia impairs vascular function and is a putative risk factor for cardiovascular and cerebrovascular diseases in underprivileged malnutrition population. The HHcy downregulates nitric oxide (NO) bioavailability by upregulating endogenous asymmetric dimethylarginine (ADMA) (Singh et al., 2009) which is an endogenous endothelial nitric oxide synthase (eNOS) inhibitor, thus results in vascular endothelial dysfunction. High levels of homocysteine may lead to increased production of oxidation products, homocysteine thiolactone and homocysteine mixed disulfides which may damage endothelium by excessive sulfation of connective tissue (Obeid and Herrmann, 2006).
Homocysteine damage the vascular endothelium which is vital part of all blood vessels performing major functions including the control of microvascular permeability, coagulation, inflammation, vascular tone as well as the formation of new vessels via vasculogenesis and angiogenesis in normal and diseased states, respectively (Endemann and Schiffrin, 2004). Damage of the blood vessels leads to Vascular Endothelial Dysfunction (VED). It is characterized by shift of endothelial actions towards reduced vasodilation, proinflammatory and prothrombic properties (Potenza et al., 2009). The modulation of vascular L-arginine/nitric oxide synthase system is hallmark of vascular endothelial dysfunction. The VED results in reduced activation of endothelium nitric oxide synthase (eNOS) and increased production of reactive oxygen species (ROS) which account for reduced synthesis and bioavailability of nitric oxide (Ulker et al., 2003) which is required for the proper working of blood vessels. The dysregulation of endothelial function upregulates the expression of procoagulant, prothrombotic and pro-inflammatory mediators. In the milieu of cardiovascular risk factors that disturb vascular homeostasis, the endothelium becomes dysfunctional resulting in enhanced production of cytokines and expression of cellular adhesion molecules by the endothelium. Various experimental evidences revealed that reduced nitric oxide production and increased oxidative stress lead to VED.
Source and generation of homocysteine: Homocysteine is generated by intra hepatic transmethylation of dietary methionine which is obtained from eggs, sesame seeds, Brazil nuts, fish, meats andcerealgrains (Ingenbleek, 2011). Methionine is converted to the methyl donor S-adenosylmethionine (SAM) by an enzyme methionine adenosyltransferase. The SAM is demethylated to S-adenosylhomocysteine (SAH) by an enzyme SAM-methyltransferase. The SAH is subsequently cleaved into adenosine and homocysteine (De Koning et al., 2003) (Fig. 1).
Metabolism of homocysteine: The metabolism of homocysteine involves two pathways i.e., remethylation pathway and transsulfuration pathway (Ingenbleek, 2011). Normally, either homocysteine is converted into methionine following remethylation (RM) pathways by transfer of a methyl group from 5-methyl tetra hydrofolate catalyzed by methionine synthase, an enzyme that requires vitamin B12 as a cofactor, to form methionine (Pandey and Pradhan, 2010) or is catabolized along the transsulfuration cascade involving the irreversible conversion of homocysteine to cysteine (Ingenbleek, 2011; Selhub, 1999). Transsulfuration reaction involves cystathionine β-synthase (CβS), an enzyme dependent on vitamin B6 (pyridoxal-5-phosphate) that condenses Hcy with serine to form cystathionine (CTT) which is further converted into cysteine through vitamin B6 (pyridoxal-5-phosphate) dependent cystathionine-γ-lyase (CγL). Cysteine, thus formed generate various products like glutathione, an important intracellular antioxidant and hydrogen sulfide (H2S), a gas that can induce endothelial-dependent relaxation and is also converted into sulfates which are excreted in urine. Cysteine can be further converted to cysteine sulfinic acid which is then converted to taurine. Taurine can decrease methionine concentration by decreasing its uptake from intestine, thus normalizing hyperhomocysteinemia. Taurine also reduces the hyperhomocysteinemia induced oxidative stress and also restores the function of extracellular superoxide dismutase thus ameliorate coronary artery wall’s pathology by its favorable effect on total plasma homocysteine and apoptosis (Wesseling et al., 2009). Excess of homocysteine is also remethylated by the enzyme betaine homocysteine methyltransferase (BHMT) which transfers a methyl group to homocysteine via demethylation of trimethylglycine (betaine) to dimethylglycine (De Koning et al., 2003) (Fig. 1).
Causes of hyperhomocysteinemia a metabolic anomaly
Deficiency of vitamins: B-vitamins are essential cofactors in both of these Hcy metabolizing pathways. An inadequate dietary intake of nutritional factors such as B-vitamins (Quadri et al., 2004) results in deficiency in all of these coenzymes viz., vitamin B12, folate and vitamin B6 thus leads to impaired metabolism of Hcy and elevated homocysteine level. Vitamin B6 (Pyridoxal-5-phosphate) shortage significantly triggers upstream accumulation of Hcy in biological fluid whereas folate or B12 deficiency results in downstream sequestration (Ingenbleek, 2011).
Genetic alteration of metabolic enzymes: Genetic alteration results in decrease in expression of Hcy metabolizing enzymes viz., cystathionine β-synthase (CβS), methylenetetrahydrofolate reductase (MTHFR) and Methionine Synthase (MS) that leads to hyperhomocysteinemia (Faraci and Lentz, 2004). The MTHFR is the rate-limiting enzyme in the methyl cycle and it is encoded by the MTHFR gene (Goyette et al., 1994). Mutations in this gene are associated with MTHFR deficiency that may increase the susceptibility to occlusive vascular disease,colon cancer, acute leukemia and neural tube defects (Fodinger et al., 2000). Higher homocysteine level has been found in an individual with C677T genotype than those with the normal C677C genotype (Lea et al., 2009).
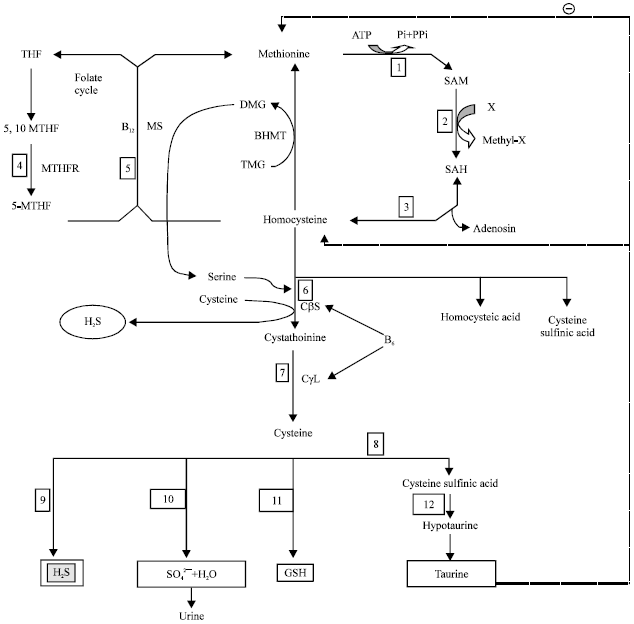 | |
| Fig. 1: | Schematic representation of the methionine cycle and homocysteine degradation pathways and their toxic metabolites, ATP: Adenosinetriphosphate, THF: Tetrahydrofolate, SAM: S-adenosylmethionine, SAH: S-adenosylhomocysteine, Cysta: Cystathionine, Cys: Cysteine, GSH: Glutathione, H2S: Hydrogen sulfide, Tau: Taurine, SO42¯: Sulfate oxyanions, TMG: Trimethylglycine and DMG: Dimethylglycine, Enzymes: (1) Met-adenosyltransferase, (2) SAM-methyltransferase, (3) Adenosyl-homocysteinase, (4) Methylene-THF reductase, (5) Met-synthase, (6) CβS indicates cystathionine-β-synthase, (7) CγL indicates cystathionine-γ-lyase, (8) oxidase, (9) CDO indicates cysteine-dioxygenase, (10) γ-glutamyl-synthase, (11) Reductase, (12) Sulfinoalanine decarboxylase, BHMT indicates betaine homocysteine methyltransferase |
The C677T polymorphism in the MTHFR enzyme leads to elevated plasma Hcy level (Sottilotta et al., 2010). The MTHFR C677T allele responsible for change in an amino acid sequence which results in diminution in MTHFR enzyme activity leads to mild hyperhomocysteinemia (Frosst et al., 1995). Both the homozygous (TT) and heterozygous (CT) genotypes are associated with lower tissue concentrations of folate, higher homocysteine concentrations and lower enzyme activity than the wild type (CC) genotype possibly resulting in vascular disease while these effects are more marked in homozygotes (Kirke et al., 2004). The human MTHFR gene mapped to chromosome 1p36.3 catalyzes the nicotinamide adenine dinucleotide phosphate (NADPH) dependent conversion of 5,10-methylenetetrahydrofolate (CH2-THF) to 5-methyltetrahydrofolate (CH3-THF), the principal circulatory form of folate and a cofactor for methylation of homocysteine to methionine. Individual with MTHFR C677T genotype, don’t effectively convert folic acid to its active form (Lea et al., 2009) and thus leads to folic acid (B-complex vitamins) deficiency with subsequent hyperhomocysteinemia which raised the risk factors for vascular dysfunction (Liu et al., 2010).
PATHOBIOLOGICAL CONSEQUENCES OF HYPERHOMOCYSTEINEMIA
Hyperhomocysteinemia and Vascular endothelium oxidative stress: The HHcy leads to the production of Reactive Oxygen Species (ROS) viz., superoxide anion (O2¯), hydroxyl radical (OH●), hydrogen peroxide (H2O2), nitric oxide free radical (NO●), peroxynitrite radical (ONOO¯) and decreases activity of antioxidants such as reduced GSH, superoxide dismutase etc. Excess Hcy leads to the formation of various ROS by following pathways:
| • | Auto oxidation pathway |
| • | Indirect pathway |
In case of autooxidation pathway, thiol group of homocysteine readily undergoes auto-oxidation in plasma to generate H2O2 free radical (ROS) (Lawrence et al., 2003).
In indirect pathway, indirect generation of O2¯ from uncoupled eNOS, xanthine oxidase, or NAD(P)H oxidase (NOX) by oxidative stress.
Excess amount of H2O2 and O2¯ (generated via autoxidation) free radicals reacts with each other in presence of Cu2+ and Fe2+ to produce OH● radical (Miller et al., 2006). The OH● radical thus formed leads to oxidation of Low Density Lipoproteine (LDL) to form lipid peroxyl radical (LOO●). Previously formed O2¯ and LOO● react with endothelium derived Nitric Oxide (NO) to produce peroxynitrites (ONOO¯) and lipidperoxynitrite (LOONO●) radicals, respectively.
ROS are potentially toxic by-products of cellular metabolism. When generated in excess, ROS can react rapidly with biomolecules causing cellular damage such as DNA mutations, lipid peroxidation and protein oxidation (Miller et al., 2006). The ROS leads to depletion of intracellular glutathione by the downregulation of antioxidant enzymes (Fig. 2).
Endogenous H2O2 free radicals generally elicits cerebral vasodilatation by opening of potassium channels while its higher concentrations causes vasoconstriction followed by vasodilatation (Miller et al., 2006).
Superoxide anion (O2¯) and hydrogen peroxide (H2O2) are known to be strong inducers of nuclear factor kappa-B (NFκ-B) activation that belong to the family of transcription factors regulate the induction and resolution of inflammation (Sharma and Singh, 2012). The NFκ-B regulates the expression of genes, including those encoding adhesins and cytokines (e.g., TNF-α and interleukin-1) (Taoufiq et al., 2008) thus, thought to promote cardiovascular disorder including endothelial dysfunction through its pro-inflammatory, pro-adhesion and pro-oxidant gene transcription. Activation of NFκ-B leads to aging induced dementia as a result of upregulation of genes of pro-inflammatory enzymes. The NFκ-B may also decrease the acetylcholine activity by increasing acetylcholinesterase (AchE) which leads to cognitive deficits and vascular impairment (Sharma and Singh, 2012).
Peroxynitrite (ONOO¯) (formed by the reaction of NO● with superoxide) is increased in peripheral blood vessels during HHcy and known to activate poly (ADP-ribose) polymerase (PARP) which is an important mediator of vascular dysfunction in disease states such as diabetes (Faraci and Lentz, 2004). The PARP activation may promote the activation of NF-κB, MAP kinases and the expression of proinflammatory mediators, adhesion molecules such as ICAM-1 and iNOS (Virag and Szabo, 2002) which subsequently effects vascular endothelial cells function. Peroxynitrite also inhibits PI3K that further reduces eNOS phosphorylation, therefore, lowering NO bioavailability which is the major consequences of vascular endothelial dysfunction (Bao et al., 2010) (Fig. 3).
Additionally, Peroxynitrite (ONOO¯) can also produce vascular endothelial dysfunction and oxidative stress by nitration of tyrosine residues in the mitochondrial isoform of superoxide dismutase (Mn-SOD) and by possibly promoting the uncoupling of NO synthases, a circumstance in which the normal flow of electrons within NO synthase is diverted to produce superoxide anion (O2¯) rather than NO (Faraci and Lentz, 2004).
Another effect of HHcy has been reported to increase in ROS production in endothelial cells of blood vessels through NADPH oxidase activation (Carluccio et al., 2007). The HHcy increased NADPH oxidase activity leads to the increase in intracellular superoxide. The NADPH oxidase (Nox) is a transmembrane/cytosolic multi-subunit enzyme that transfers electrons from NADPH to molecular oxygen to produce superoxide.
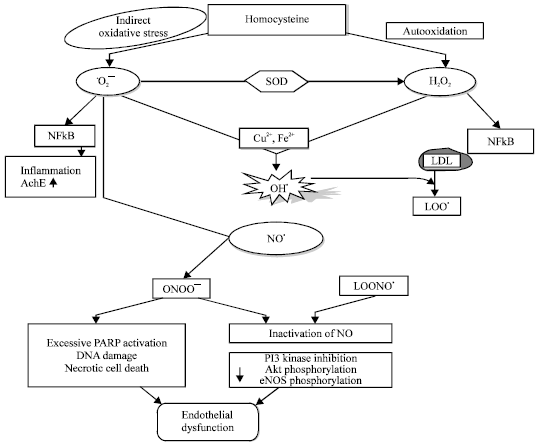 | |
| Fig. 2: | Homocysteinl and oxidative stress related endothelial dysfunction, Superoxide anion (O2¯), Hydrogen peroxide (H2O2), Hydroxyl radical (OH●), Lipidperoxyl radical (LOO●), Nitric oxide free radical (NO●), Peroxynitrite radical (ONOO¯), Lipid peroxynitrite radical (LOONO.), Superoxide dismutase (SOD), Hydrogen sulfide (H2S), Nuclear factor kappa-B (NFκ-B), Acetylcholinesterase (AchE) |
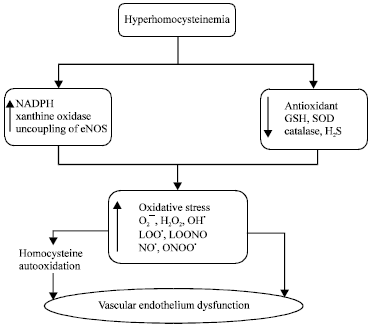 | |
| Fig. 3: | Hyperhomocysteinemia and oxidative stress related endothelial dysfunction, O2¯: Superoxide anion, H2O2: Hydrogen peroxide, OH●: Hydroxyl radical, LOO●: Lipidperoxyl radical, LOONO¯: Lipid peroxynitrite radical; NO●: Nitric oxide free radical, ONOO¯: Peroxynitrite radical, GSH: Reduced glutathione, SOD: Superoxide dismutase, H2S: Hydrogen sulfide, NFκ-B: Nuclear factor kappa-B |
Superoxide produced by Nox can spontaneously form hydrogen peroxide (H2O2) which undergoes further reactions to generate ROS. However, Nox is composed of multiple cytosolic regulatory components (p47phox, p67phox, p40phox and rac) and a transmembrane catalytic heterodimer (p22phox and gp91phox). When Nox is activated, regulatory cytosolic components are translocated to cellular membranes, where they form a functional complex that generates superoxide radicals. The Nox family of NADPH oxidases has seven members, including Nox1, Nox2, Nox3, Nox4, Nox5, Duox1 and Duox2 (Bao et al., 2009). The NADPH oxidase (Nox4) has been located in the nucleus of endothelial cells and it has been suggested that it might regulate gene expression and activate a serine/threonine protein kinase through the production of ROS (Bao et al., 2009).
Moreover, HHcy increases TNF-α expression which enhances oxidative stress through upregulating a Nox1 based NAD(P)H oxidase. Thus, TNF-α induces a proinflammatory vascular phenotype in HHcy that potentially contributes to the development of coronary atherosclerosis (Ungvari et al., 2003). It is hypothesized that in HHcy, TNF-α upregulates NAD(P)H oxidase-dependent O2¯ production both by induction of the oxidase subunit Nox1 and an increased phosphorylation of the regulatory subunits. HHcy also induces VCAM-1 gene expression through NAD(P)H oxidase activation. The VCAM-1 expression is crucially involved in the monocyte adhesion and early atherogenesis in the vascular wall (Carluccio et al., 2007). Excess of Hcy decreases the binding of extracellular superoxide dismutase (EC-SOD) to vascular endothelial cell surfaces by degradation of endothelial heparan sulfate proteoglycan which results in a loss of the ability to protect endothelial cell surfaces from oxidative stress. The EC-SOD in the vascular system diffuses into the extracellular space and scavenges superoxide before it reacts with No. Elimination of EC-SOD from the endothelium may result in the elevation of superoxide which would then produce highly reactive peroxynitrite (ONOO¯) (Yamamoto et al., 2000). Therefore, it has been suggested that HHcy decreases the ability to scavenge superoxide and indirectly causes injury of the vascular endothelium.
HHcy also impairs the glutathione peroxidase activity (Handy et al., 2005) thus sensitizing cells to the cytotoxic effects of agents or conditions known to generate ROS. Catalase or glutathione peroxidase normally metabolizes H2O2 into water. So decrease in activity of these enzymes may lead to non-enzymatic conversion of H2O2 into OH• by reaction with heavy metals (Fe2+) (Miller et al., 2006) (Fig. 2).
Furthermore, oxidative stress accounts for the initiation of apoptosis and the dysfunction of EPCs induced by HHcy. Impairment of NO-mediated vascular protection is also related to the effects of Hcy on endothelial progenitor cells (EPCs) (Bao et al., 2010).
Hyperhomocysteinemia and vascular endothelium interacellular pathways: The HHcy impairs the cellular and molecular mechanisms of vascular endothelial cells by causing imbalance between phosphorylation and dephosphorylation state of lipid and protein kinases which modulates vascular L-arginine/nitric oxide synthatase activity and may produce vascular endothelium dysfunction. The HHcy has been reported to impair Phosphatidylinositide 3-kinases/Protein Kinase B (PKB) [PI3K/Akt] pathway by activation of protein tyrosine phosphatase (PTPase) (Sharma et al., 2011a) and Rho kinase mediated intracellular signaling pathway (Park et al., 2011).
Activation of PI3K/Akt pathway mediates the rapid increase in eNOS activity (Sharma et al., 2011b) thus increasing the availability of No. Overactivation of PTPase and decrease in PI3K/Akt pathway is implicated in the regulation of cellular proliferation, differentiation and apoptosis by its action to enhance proapoptotic genes such as Caspase, BCL-2 and Bcl-2-associated death promoter (BAD) (Sharma et al., 2011a). The PTPase also attenuates various growth factors or extracellular matrix (Meredith et al., 1993) and inactivates Mitogen-Activated Protein Kinases (MAP Kinases) which leads to endothelial cells apoptosis (Harrington et al., 2000). Inhibition in PTPase activity have been reported to attenuate hyperhomocysteinemia induced vascular and cardiac complications (Harrington et al., 2000).
Rho-kinase is reported to be involved in vascular endothelial dysfunction. Rho kinase is a small GTP-binding protein involved in intracellular signaling pathway. Hyperhomocysteinemia leads to abnormal activation of Rho kinase pathway (Taoufiq et al., 2008) which likely to be acts upstream of PI3K/Akt (Park et al., 2011). Rho-kinase also increases the generation of ROS (Brown et al., 2006) which reduces the biosynthesis of NO (Kumar et al., 2007; Nohria et al., 2006). Rho kinase also increases the vascular endothelium permeability (Clements et al., 2005) by modulating the phosphorylation of myosin light chain as well as tight junctions (Taoufiq et al., 2008). Rho kinase can also activate various cascades through its ability to phosphorylate multiple substrates (including other kinases), thereby increasing NFκ-B activity (Taoufiq et al., 2008) that leads to vascular impairment. Inhibition of Rho-kinase by fasudil leads to the rapid phosphorylation and activation of Akt via PI3K, resulting in an increase in NO production (Sharma et al., 2011b) and improving hyperhomocysteinemia induced vascular endothelial dysfunction (Shah and Singh, 2006a).
Hyperhomocysteinemia is involved in the production of high oxidative stress which has been reported to activate Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. c-Jun N-terminal kinases (JAKs) family consists of JAK1, JAK2, JAK3 and tyrosine kinases-2 (Tyk2) (Marrero et al., 1995). Increase in activation of protein tyrosine phosphataseshas been documented to remove phosphates from cytokine receptors and activate STATs (Hebenstreit et al., 2005). Thus, activated JAK pathway has been demonstrated to inhibit PI3K/AKT pathway (Faraci and Lentz, 2004).
Proteine Kinase A (PKA) activation has been reported to improve hyperhomocysteinemia induced vascular endothelium dysfunction (VED) by modulating various intracellular pathways. It is reported that 8-Br-cAMP, an activator of PKA ameliorates hyperhomocysteinemia induced VED. The cAMP-dependent protein kinase A is the principle intracellular target for cAMP in mammalian cells (Shah and Singh, 2006b). The PKA activation stimulates phosphorylation of eNOS and opening of ATP sensitive K+ channels which leads to activation of eNOS and thus increases the bioavailability of NO (Boo and Jo, 2003). The PKA also attenuate Rho-kinase associated inhibition of eNOS via PI3K/Akt pathway (Kumar et al., 2007; Boo and Jo, 2003), PKA also reduces the hyperhomocysteinemia induced generation of ROS (Sullivan et al., 2001) and mediates the Bradykinin induced production of endothelial NO (Bae et al., 2003) thereby, decreasing the permeability of endothelium and apoptotic death of endothelial cells (Irie et al., 2001; Kulhanek-Heinze et al., 2004) thus ameliorating hyperhomocysteinemia related vascular endothelial dysfunction (Fig. 4).
Hyperhomocysteinemia and vascular endothelium progenitor cells: Progenitor cells are primitive Bone Marrow (BM) cells that are located at the periphery of the blood islands and have the capacity to proliferate, migrate and differentiate into mature endothelial lineage cells (Asahara and Kawamoto, 2004). Endothelial Progenitor Cells (EPCs) are derived from more differentiated CD34 or immature CD133 hematopoietic stem cells and blood mononuclear cells or CD14 monocytes. The EPCs maintain vascular homeostasis by restoring an intact endothelium (Kruman et al., 2000). The EPCs provides a circulating pool of cells that could form a cellular patch at the site of denuding injury and thus act as cellular reservoir to eplace dysfunctional endothelium. Impaired mobilization or depletion of these EPCs contributes to endothelial dysfunction (Hill et al., 2003).
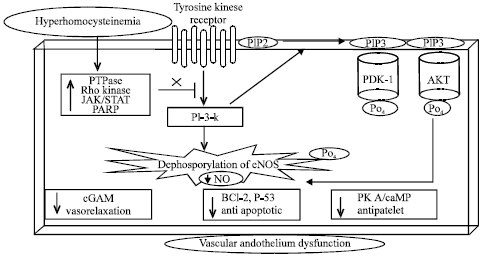 | |
| Fig. 4: | Vascular endothelium cell intracellular PI3K/PDK/Akt pathway attenuated by hyperhomocysteinemia due to increase in protein tyrosine phosphatase, Rho kinase, JAK/STAT and PARP that leads to vascular endothelium dysfunction. PTPase: Protein tyrosine phosphatase, JAK/STAT: Janus kinase/signal transducer and activator of transcription, PARP: Poly (ADP-ribose) polymerase, PI3K: Phosphatidylinositide 3-kinases, Akt: Protein Kinase B (PKB), PIP2: Phosphatidylinositol 4,5-bisphosphate, PDK-1: Phosphoinositide dependent protein kinase-1, cGMP: Cyclic guanosine monophosphate, PKA: Protein kinase A |
Hyperhomocysteinemia (HHcy) reduces Endothelial Progenitor Cell (EPC) numbers and impairs its functional activity (Zhu et al., 2006) which is associated with EPCs senescence. The HHcy accelerates the onset of EPCs senescence through telomerase inactivation or by decreasing the Akt phosphorylation leading to cellular dysfunction (Zhu et al., 2006; Xia et al., 2008). Telomerase acts by delay or decreasing endothelial cell senescence by Akt dependent manner. Akt acts by increasing the activity of the eNOS, thereby, increasing the availability of NO that has been demonstrated to activate telomerase and reprieves endothelial cell senescence. The HHcy mediated EPCs apoptosis is also associated with caspase-8, cytochrome-c release and caspase-3 activation. HHcy induced increased ROS, acting as an upstream factor for mitochondrial membrane depolarization, forces mitochondria to release cytochrome-c and caspases, resulting in eventual cellular apoptosis and dysfunction (Bao et al., 2010) (Fig. 5).
Immune activation, Hyperhomocysteinemia and vascular endothelium proinflammatory mediators: One of the major cause of HHcy is the activation of immune system. Most diseases associated with hyperhomocysteinemia are due to immune system activation (Erren et al., 1999; Rogers et al., 1996). Chronic activation of the immune system leads to the enhanced production of oxidative stress which may be the major pathogenic mechanisms contributing to the development and progression of diseases such as atherosclerosis, vascular dementia etc. Many factors such as aging, injury, cancer, autoimmunity etc. are reported in literature that stimulates immune system which further activates immunocompetent T-cells and monocytes. Proliferated T-cells (Th1-type immune activation) release Interferon-γ (IFN-γ) and Hcy. The IFN-γ further stimulates monocytes to release ROS (Schroecksnadel et al., 2003) whereas, Hcy get accumulated and start cascade of producting ROS. ROS thus formed activates nuclear factor kappa-B (NFk-B) which enhances cytokine production eg. tumor necrosis factor-α (TNF-α) (Lazzerini et al., 2007) and also upregulates the expression of various adhesion molecules such as vascular cell adhesion molecule (VCAM-1) and E-selectin, P-selectin (Silverman et al., 2002) with subsequent inflammatory responses and endothelial dysfunction (Pruefer et al., 1999) (Fig. 5). The NFk-B activation modifies the expression of Monocyte Chemoattractant Protein-1 (MCP-1) (Wang et al., 2000). MCP-1 is a potent chemokine (Nelken et al., 1991) that together with adhesion (molecules VCAM-1 and E-selectin) bind to circulating monocyte/macrophage, leading to cell attachment and migration into the sub-endothelial space (Wang et al., 2002) which activates inflammatory cascade (Li and Glass, 2002). It clearly shows that HHcy is the interlink between the immune system and adhesion molecules (Schroecksnadel et al., 2003).
Enhanced ROS also initiates oxidation of Low-Density Lipoprotein (LDL) to produce Modified Low Density Lipoprotein (MLDL). These MLDL is taken up by macrophages and ending up with the formation of foam cells (Schroecksnadel et al., 2003).
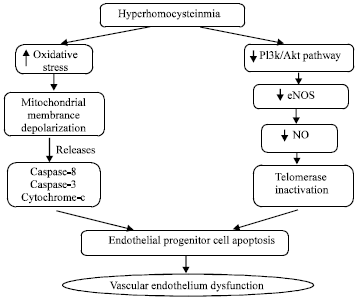 | |
| Fig. 5: | Endothelial progenitor cell apoptosis and vascular endothelium dysfunction by hyperhomocysteinemia |
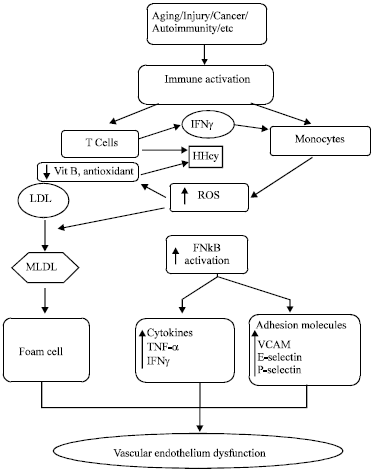 | |
| Fig. 6: | Hyperhomocysteinemia and immune cell activation in the development of atherosclerosis. ROS: Reactive oxygen species, LDL: Low-density lipoprotein, MLDL: Modified low-density lipoprotein, NFk-B: Nuclear factor kappa-B, TNF-α: Tumor necrosis factor-α, INF-γ: Interferon gamma, VCAM: Vascular cell adhesion molecule |
Foam cells are not dangerous as such but can become a problem when they accumulate at particular site of the blood vessels. It creates a necrotic centre ofatherosclerosis which obstructs the adequate amount of blood to flow resulting inischemia and contributing to stroke and myocardial infarction which are the two leading causes of cardiovascular death related to hyperhomocysteinemia (Fig. 6).
Hyperhomocysteinemia, red blood cells and vascular endothelium dysfunction: The 5-MTHF transfers its methyl group to Hcy and converted into methionine and THF (Fig. 1). This reaction requires vit B12 as a cofactor. Deficiency of vit B12 and mutation in enzyme MTHFR C677T allele (Ventura et al., 2004) leads to accumulation of Hcy which further causes the RBCs haemolysis.
Homocysteine has been proposed as a hemolytic toxin (Acharya et al., 2008). The HHcy cause vascular endothelium dysfunction consequence of which leads to immediate microangiopathy (A disease of the capillaries) initiating the lysis of the RBCs (Acharya et al., 2008). The HHcy activates polymorphonuclear leukocytes which increases hemolytic activity of RBCs (Olinescu et al., 1996). It has been well documented in literature that homocysteine down regulates the cellular glutathione peroxidase-1 activity that increases the production of reactive oxygen species which may subsequently provoke the oxidative vulnerability of sulfhydryl groups of hemoglobin leading to hemoglobin precipitation within the RBC and induces cell lysis.
Hyperhomocysteinemia also stimulates NMDA receptors (present on the erythrocytic membrane) which induce the Ca2+ intake in the RBCs (Boldyrev, 2010) thus leads to RBCs aggregation and structural deformation causing hemolysis (Muravyov and Tikhomirova, 2012). Moreover, infected erythrocytes adhere to the endothelial cells can directly trigger the Rho kinase activity thereby may block the PI3K/Akt pathway mediated eNOS activation (Taoufiq et al., 2008) and results in increased production of Reactive Oxygen Species (ROS) by endothelial cells, leading to vascular endothelium dysfunction or cell death (Pino et al., 2003; Taoufiq et al., 2006) (Fig. 7).
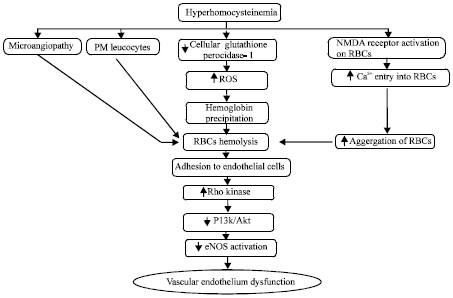 | |
| Fig. 7: | Hyperhomocysteinemia induced vascular nedothelium dysfunction by RBC hemolysis. PMN: Polymrophonuclear |
CONCLUSION
It has been clear that mutation in MTHFR genes, deficiency of vitamin B12, folate, betaine and B6 as a result of malnutrition, normal aging and diseased conditions are the main causes for increased homocysteine level. Despite the fact that there have been major advances in the identification of HHcy as an important independent metabolic anomaly, molecular regulators in disease progression and its role in the various pathological conditions remains to be identified. Better understanding of pathology of HHcy may provide interventions in the regulation and progression of several vascular disorders such as dementia, ischemia, cardiovascular diseases and haemostatic disorders. Vascular endothelium dysfunction is the first event that is followed by smooth muscle and basement membrane abnormalities that leads to insufficient blood supply and exaggerate vascular pathology. Thus further studies are warranted to translate this scientific knowledge into potential pharmacological interventions for HHcy and its associated vascular disorders.
REFERENCES
- Acharya, U., J.T. Gau, W. Horvath, P. Ventura, C.T. Hsueh and W. Carlsen, 2008. Hemolysis and hyperhomocysteinemia caused by cobalamin deficiency: Three case reports and review of the literature. J. Hematol. Oncol., Vol. 1.
CrossRef - Asahara, T. and A. Kawamoto, 2004. Endothelial progenitor cells for postnatal vasculogenesis. Am. J. Physiol. Cell Physiol., 287: C572-C579.
CrossRef - Bae, S.W., H.S. Kim, Y.N. Cha, Y.S. Park, S.A. Jo and I. Jo, 2003. Rapid increase in endothelial nitric oxide production by bradykinin is mediated by protein kinase A signaling pathway. Biochem. Biophys. Res. Commun., 306: 981-987.
CrossRef - Bao, X.M., C.F. Wu and G.P. Lu, 2009. Atorvastatin attenuates homocysteine-induced apoptosis in human umbilical vein endothelial cells via inhibiting NADPH oxidase-related oxidative stress-triggered p38MAPK signaling. Acta Pharmacol. Sin., 30: 1392-1398.
CrossRef - Bao, X.M., C.F. Wu and G.P. Lu, 2010. Atorvastatin inhibits homocysteine-induced dysfunction and apoptosis in endothelial progenitor cells. Acta Pharmacol. Sin., 31: 476-484.
CrossRef - Boo, Y.C. and H. Jo, 2003. Flow-dependent regulation of endothelial nitric oxide synthase: Role of protein kinases. Am. J. Physiol. Cell Physiol., 285: C499-C508.
CrossRef - Brown, J.H., D.P. del Re and M.A. Sussman, 2006. The Rac and Rho hall of fame a decade of hypertrophic signaling hits. Circulation Res., 98: 730-742.
CrossRefDirect Link - Carluccio, M.A., M.A. Ancora, M. Massaro, M. Carluccio and E. Scoditti et al., 2007. Homocysteine induces VCAM-1 gene expression through NF-κB and NAD (P) H oxidase activation: Protective role of Mediterranean diet polyphenolic antioxidants. Am. J. Physiol.-Heart Circulatory Physiol., 293: H2344-H2354.
CrossRefDirect Link - Clements, R.T., F.L. Minnear, H.A. Singer, R.S. Keller and P.A. Vincent, 2005. RhoA and Rho-kinase dependent and independent signals mediate TGF-β-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am. J. Physiol. Lung Cell. Mol. Physiol., 288: L294-L306.
CrossRefDirect Link - Dalton, M.L., P.F. Gadson, R.W. Wrenn and T.H. Rosenquist, 1997. Homocysteine signal cascade: Production of phospholipids, activation of protein kinase C and the induction of c-fos and c-myb in smooth muscle cells. FASEB. J., 11: 703-711.
Direct Link - Erren, M., H. Reinecke, R. Junker, M. Fobker and H. Schulte et al., 1999. Systemic inflammatory parameters in patients with atherosclerosis of the coronary and peripheral arteries. Arteriosclerosis Thrombosis Vascular Biol., 19: 2355-2363.
CrossRef - Faraci, F.M. and S.R. Lentz, 2004. Hyperhomocysteinemia, oxidative stress and cerebral vascular dysfunction. Stroke, 35: 345-347.
CrossRefDirect Link - Fodinger, M., W.H. Horl and G. Sunder-Plassmann, 2000. Molecular biology of 5, 10-methylenetetrahydrofolate reductase. J. Nephrol., 13: 20-33.
PubMedDirect Link - Frosst, P., H.J. Blom, R. Milos, P. Goyette and C.A. Sheppard et al., 1995. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet., 10: 111-113.
CrossRefDirect Link - Goyette, P., J.S. Summer and R. Milos, 1994. Human methylenetetrahydrofolate reductase: Isolation of cDNA, mapping and mutation. Nat. Genet., 7: 195-200.
Direct Link - Handy, D.E., Y. Zhang and J. Loscalzo, 2005. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J. Biol. Chem., 280: 15518-15525.
CrossRefDirect Link - Harrington, E.O., A. Smeglin, N. Parks, J. Newton and S. Rounds, 2000. Adenosine induces endothelial apoptosis by activating protein tyrosine phosphatase: A possible role of p38α. Am. J. Physiol. Lung Cell. Mol. Physiol., 279: L733-L742.
Direct Link - Hebenstreit, D., J. Horejs-Hoeck and A. Duschl, 2005. JAK/STAT-dependent gene regulation by cytokines. Drug News Perspect, 18: 243-249.
Direct Link - Hill, J.M., G. Zalos, J.P.J. Halcox, W.H. Schenke, M.A. Waclawiw, A.A. Quyyumi and T. Finkel, 2003. Circulating endothelial progenitor cells, vascular function and cardiovascular risk. N. Engl. J. Med., 348: 593-600.
CrossRefDirect Link - Ingenbleek, Y., 2011. The oxidative stress of hyperhomocysteinemia results from reduced bioavailability of sulfur-containing reductants. Open Clin. Chem. J., 4: 34-44.
Direct Link - Irie, K., E. Fujii, H. Ishida, K. Wada and T. Suganuma et al., 2001. Inhibitory effects of cyclic AMP elevating agents on lipopolysaccharide (LPS)‐induced microvascular permeability change in mouse skin. Br. J. Pharmacol., 133: 237-242.
CrossRef - Kirke, P.N., J.L. Mills, A.M. Molloy, L.C. Brody and V.B. O'Leary et al., 2004. Impact of the MTHFR C677T polymorphism on risk of neural tube defects: Case-control study. BMJ, 328: 1535-1536.
CrossRefDirect Link - Kruman, I.I., C. Culmsee, S.L. Chan, Y. Kruman, Z. Guo, L. Penix and M.P. Mattson, 2000. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci., 20: 6920-6926.
Direct Link - Kulhanek-Heinze, S., A.L. Gerbes, T. Gerwig, A.M. Vollmar and A.K. Kiemer, 2004. Protein kinase A dependent signalling mediates anti-apoptotic effects of the atrial natriuretic peptide in ischemic livers. J. Hepatol., 41: 414-420.
CrossRef - Kumar, R., V.P. Singh and K.M. Baker, 2007. Kinase inhibitors for cardiovascular disease. J. Mol. Cell. Cardiol., 42: 1-11.
CrossRef - De Koning, A.B.L., G.H. Werstuck, J. Zhou and R.C. Austin, 2003. Hyperhomocysteinemia and its role in the development of atherosclerosis. Clin. Biochem., 36: 431-441.
CrossRef - Lazzerini, P.E., P.L. Capecchi, E. Selvi, S. Lorenzini, S. Bisogno, M. Galeazzi and F. Laghi Pasini, 2007. Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmunity Rev., 6: 503-509.
CrossRef - Lea, R., N. Colson, S. Quinlan, J. Macmillan and L. Griffiths, 2009. The effects of vitamin supplementation and MTHFR (C677T) genotype on homocysteine-lowering and migraine disability. Pharm. Genomics, 19: 422-428.
CrossRefDirect Link - Li, A.C. and C.K. Glass, 2002. The macrophage foam cell as a target for therapeutic intervention. Nat. Med., 8: 1235-1242.
Direct Link - Liu, A., S. Menon, N.J. Colson, S. Quinlan and H. Cox et al., 2010. Analysis of the MTHFR C677T variant with migraine phenotypes. BMC Res. Notes, Vol. 3.
CrossRef - Meredith, J.E.J., B. Fazeli and M.A. Schwartz, 1993. The extracellular matrix as a cell survival factor. Mol. Biol. Cell, 4: 953-961.
Direct Link - Miller, A.A., G.R. Drummond and C.G. Sobey, 2006. Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol. Ther., 111: 928-948.
CrossRef - Muravyov, A. and I. Tikhomirova, 2012. Role Ca2+ in mechanisms of the red blood cells microrheological changes. Adv. Exp. Med. Biol., 740: 1017-1038.
CrossRef - Nelken, N.A., S.R. Coughlin, D. Gordon and J.N. Wilcox, 1991. Monocyte chemoattractant protein-1 in human atheromatous plaques. J. Clin. Invest., 88: 1121-1127.
CrossRefDirect Link - Nohria, A., M.E. Grunert, Y. Rikitake, K. Noma and A. Prsic et al., 2006. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circulation Res., 99: 1426-1432.
CrossRef - Obeid, R. and W. Herrmann, 2006. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett., 580: 2994-3005.
CrossRef - Olinescu, R., F.A. Kummerow, B. Handler and L. Fleischer, 1996. The hemolytic activity of homocysteine is increased by the activated polymorphonuclear leukocytes. Biochem. Biophys. Res. Commun., 226: 912-916.
CrossRef - Pandey, P. and S. Pradhan, 2010. Homocysteine: A possible modifiable risk factor in vascular dementia. Ann. Neurosci., 13: 12-17.
Direct Link - Park, H.J., Y.H. Choi, Y.J. Cho, P.M. Henson and J.L. Kang, 2011. RhoA-mediated signaling up-regulates hepatocyte growth factor gene and protein expression in response to apoptotic cells. J. Leukocyte Biol., 89: 399-411.
CrossRefDirect Link - Pino, P., I. Vouldoukis, N. Dugas, G. Hassani‐Loppion, B. Dugas and D. Mazier, 2003. Redox‐dependent apoptosis in human endothelial cells after adhesion of Plasmodium falciparum-infected erythrocytes. Ann. N. Y. Acad. Sci., 1010: 582-586.
CrossRef - Potenza, M.A., S. Gagliardi, C. Nacci, M.R. Carratu and M. Montagnani, 2009. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem., 16: 94-112.
CrossRef - Pruefer, D., R. Scalia and A.M. Lefer, 1999. Homocysteine provokes leukocyte-endothelium interaction by downregulation of nitric oxide. General Pharmacol.: Vascular Syst., 33: 487-498.
CrossRef - Quadri, P., C. Fragiacomo, R. Pezzati, E. Zanda, G. Forloni, M. Tettamanti and U. Lucca, 2004. Homocysteine, folate and vitamin B-12 in mild cognitive impairment, Alzheimer disease and vascular dementia. Am. J. Clin. Nutr., 80: 114-122.
Direct Link - Rogers, J., S. Webster, L.F. Lue, L. Brachova and W.H. Civin et al., 1996. Inflammation and Alzheimer's disease pathogenesis. Neurobiol. Aging, 17: 681-686.
CrossRefPubMedDirect Link - Schroecksnadel, K., B. Frick, C. Winkler, F. Leblhuber, B. Wirleitner and D. Fuchs, 2003. Hyperhomocysteinemia and immune activation. Clin. Chem. Lab. Med., 41: 1438-1443.
CrossRefDirect Link - Shah, D.I. and M. Singh, 2006. Activation of protein kinase A improves vascular endothelial dysfunction. Endothelium, 13: 267-277.
Direct Link - Shah, D.I. and M. Singh, 2006. Effect of fasudil on macrovascular disorder-induced endothelial dysfunction. Can. J. Physiol. Pharmacol., 84: 835-845.
CrossRefDirect Link - Sharifi, F., H. Fakhrzadeh, M. Mirarefin, R. Pourebrahim and M. Nouri et al., 2010. The effects of high-dose folic acid on blood pressure of hypertensive adults with hyperhomocysteinemia: A randomized double-blind placebo controlled clinical trial (Tehran Homocysteine survey). J. Diabetes Metabolic Disorders, 9: 1-13.
- Sharma, B. and N. Singh, 2012. Salutary effect of NFκB inhibitor and folacin in hyperhomocysteinemia-hyperlipidemia induced vascular dementia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 38: 207-215.
CrossRef - Sharma, S., M. Singh and P.L. Sharma, 2011. Beneficial effect of insulin in hyperhomocysteinemia and diabetes mellitus-induced vascular endothelium dysfunction: Role of phosphoinositide dependent kinase and protein kinase B. Mol. Cell. Biochem., 348: 21-32.
CrossRef - Sharma, S., M. Singh and P.L. Sharma, 2011. Mechanism of attenuation of diabetes mellitus and hypercholesterolemia induced vascular endothelial dysfunction by protein tyrosine phosphatase inhibition. Vasc. Pharmacol., 54: 80-87.
CrossRef - Silverman, M.D., R.J. Tumuluri, M. Davis, G. Lopez, J.T. Rosenbaum and P.I. Lelkes, 2002. Homocysteine upregulates vascular cell adhesion molecule-1 expression in cultured human aortic endothelial cells and enhances monocyte adhesion. Arteriosclerosis Thrombosis Vasc. Biol., 22: 587-592.
CrossRefDirect Link - Sottilotta, G., S.M. Siboni, C. Latella, V. Oriana and E. Romeo et al., 2010. Hyperhomocysteinemia and C677T MTHFR genotype in patients with retinal vein thrombosis. Clin. Applied Thrombosis/Hemostasis, 16: 549-553.
CrossRefDirect Link - Sullivan, G.W., J.M. Rieger, W. Michael Scheld, T.L. Macdonald and J. Linden, 2001. Cyclic AMP-dependent inhibition of human neutrophil oxidative activity by substituted 2-propynylcyclohexyl adenosine A2A receptor agonists. Br. J. Pharmacol., 132: 1017-1026.
CrossRef - Taoufiq, Z., P. Pino, N. Dugas, M. Conti, M. Tefit, D. Mazier and I. Vouldoukis, 2006. Transient supplementation of superoxide dismutase protects endothelial cells against Plasmodium falciparum-induced oxidative stress. Mol. Biochem. Parasitol., 150: 166-173.
CrossRef - Ulker, S., P.P. McKeown and U. Bayraktutan, 2003. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension, 41: 534-539.
CrossRefDirect Link - Ungvari, Z., A. Csiszar, J.G. Edwards, P.M. Kaminski, M.S. Wolin, G. Kaley and A. Koller, 2003. Increased superoxide production in coronary arteries in hyperhomocysteinemia: Role of tumor necrosis factor-α, NAD(P)H oxidase and inducible nitric oxide synthase. Arteriosclerosis Thrombosis Vasc., 23: 418-424.
CrossRefPubMedDirect Link - Ventura, P., R. Panini, S. Tremosini and G. Salvioli, 2004. A role for homocysteine increase in haemolysis of megaloblastic anaemias due to vitamin B12 and folate deficiency: results from an in vitro experience Biochimica Biophysica Acta (BBA)-Mol. Basis Dis., 1739: 33-42.
CrossRefDirect Link - Virag, L. and C. Szabo, 2002. The therapeutic potential of poly (ADP-ribose) polymerase inhibitors. Pharmacol. Rev., 54: 375-430.
Direct Link - Wang, G., C.W. Woo, F.L. Sung, Y.L. Siow and O. Karmin, 2002. Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia role of chemokine and adhesion molecules. Arteriosclerosis Thrombosis Vasc. Biol., 22: 1777-1783.
CrossRefDirect Link - Wang, G., Y.L. Siow and O. Karmin, 2000. Homocysteine stimulates nuclear factor κB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: A possible role for protein kinase C. Biochem. J., 352: 817-826.
Direct Link - Wesseling, S., M.P. Koeners and J.A. Joles, 2009. Taurine red bull or red herring? Hypertension, 53: 909-911.
CrossRef - Xia, L., X.X. Wang, X.S. Hu, X.G. Guo and Y.P. Shang et al., 2008. Resveratrol reduces endothelial progenitor cells senescence through augmentation of telomerase activity by Akt-dependent mechanisms. Br. J. Pharmacol., 155: 387-394.
CrossRef - Yamamoto, M., H. Hara and T. Adachi, 2000. Effects of homocysteine on the binding of extracellular-superoxide dismutase to the endothelial cell surface. FEBS Lett., 486: 159-162.
CrossRef - Zhu, J.H., J.Z. Chen, X.X. Wang, X.D. Xie, J. Sun and F.R. Zhang, 2006. Homocysteine accelerates senescence and reduces proliferation of endothelial progenitor cells. J. Mol. Cell. Cardiol., 40: 648-652.
CrossRef - Lawrence, A., C.M. Jones, P. Wardman and M.J. Burkitt, 2003. Evidence for the role of a peroxidase compound I-type intermediate in the oxidation of glutathione, NADH, ascorbate and dichlorofluorescin by cytochrome c/H2O2. Implications for oxidative stress during apoptosis. J. Biol. Chem., 278: 29410-29419.
PubMed