
Yun Fu
Department of Molecular Biology and Biochemistry, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Yu Zhang
Department of Molecular Biology and Biochemistry, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Sufeng Zhou
Clinical Skill Training Center, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Youxun Liu
Department of Molecular Biology and Biochemistry, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Jiangang Wang
Department of Physiology, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Yali Wang
Department of Physiology, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Chengbiao Lu
Department of Physiology, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
Changzheng Li
Department of Molecular Biology and Biochemistry, Xinxiang Medical University, Xinxiang,Henan, 453003, People�s Republic of China
International Journal of Pharmacology
Year: 2013 | Volume: 9 | Issue: 7 | Page No.: 416-429







ABSTRACT
Ciprofloxacin is one of fluoroquuinones widely used as antibiotics in clinical treatments. It has been shown that the carboxyl group on position 3 of ciprofloxacin is essential for antimicrobial activity, however, the amide form on this position and its corresponding biological effects have not been studied. To determine the structure-activity relationship, the ciprofloxacin hydrazide derivative was synthesized and its antimicrobial and antitumor mechanism was also evaluated preliminarily. Our results demonstrated that the substitution of -OH of carboxyl on position 3 of ciprofloxacin with a -NH-NH2 group could slightly alter the antimicrobial spectra, but not significantly. The studies of the hydrazide derivative on cell cycle arrest, mitochondrial membrane permeability, topoisomerase inhibition, pro-apoptotic gene regulation and molecular docking revealed that the minor structural modification on position 3 of ciprofloxacin did not result in changes in molecular targets compared to ciprofloxacin.
PDF Abstract XML References Citation
Received: January 24, 2014;
Accepted: March 17, 2014;
Published: April 15, 2014
How to cite this article
Yun Fu, Yu Zhang, Sufeng Zhou, Youxun Liu, Jiangang Wang, Yali Wang, Chengbiao Lu and Changzheng Li, 2013. Effects of Substitution of Carboxyl with Hydrazide Group on Position 3 of Ciprofloxacin on its Antimicrobial and Antitumor Activity. International Journal of Pharmacology, 9: 416-429.
DOI: 10.3923/ijp.2013.416.429
URL: https://scialert.net/abstract/?doi=ijp.2013.416.429
DOI: 10.3923/ijp.2013.416.429
URL: https://scialert.net/abstract/?doi=ijp.2013.416.429
INTRODUCTION
The fluoroquinolones are groups of synthetic antibiotics that have their broad-spectrum antimicrobial activities against gram-negative and gram-positive bacterial pathogens via binding to topoisomerase enzymes, causing double stranded DNA breaks and finally resulting in cell death (Patitungkho et al., 2011). Some members of this family have been demonstrated to exhibit anti-tumor activity in cancer cell lines (Fabian et al., 2006; Thadepalli et al., 2005), such as colon cancer, bladder cancer and leukemia cell lines. The antitumor activity of them is related to the topoisomerase II inhibitory activity (Hawtin et al., 2010; Seo et al., 2012). Ciprofloxacin is one of the safest antimicrobial drugs, its derivatives have been synthesized (Yadav and Joshi, 2008; Lemaire et al., 2011; Jubie et al., 2010; Patitungkho et al., 2011) to improve its antimicrobial or antitumor activity. The accumulated knowledge enables us to find the Structure-activity Relationship (SAR) and now the functions of different positions on the quinolone core have been identified (Ahmed and Daneshtalab, 2012). The SAR studies indicated that position 3 and 4 of ciprofloxacin are essential for gyrase binding and bacterial transport and no other useful substitutions have yet been reported. The 3-carboxylate and 4-carbonyl groups are, therefore, considered essential for antimicrobial activity (Tillotson, 1996). Hu et al. (2012) reported a series of substitutes of s-triazolothiadiazines and pyrazolo s-triazoles on the position 3 of ciprofloxacin, with IC50 ranging from 1 to 23.8 μM for the investigated cancer cell lines (Hu et al., 2012). Devnath and Islam, fused the position 3 and 4 with pyrazolone derivatives, showing good lethality against brine shrimp (Devnath and Islam, 2010). Those studies indicated that the groups on position 3 or 4 of ciprofloxacin can be substituted by different chemical groups and the derivatives have good biological activities even though the antimicrobial activity decreased.
In the present study, the necessity of the carboxyl group on position 3 of ciprofloxacin in topoisomerase binding and bacterial transport was questioned, therefore, substituted the -OH group of carboxylate with an -NHNH2 (hydrazide) group (amide) and investigated what changes this may introduce. In the present study, we evaluated the effects of the structural modification on antimicrobial and antitumor activity, cell cycle arrest, depolarization of mitochondrial membrane and topoisomerase inhibition.
MATERIALS AND METHODS
Materials: All chemical reagents are analytical reagent grade. MTT (4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide), Rhodamine123, propidium iodide and SDS (Sodium dodecyl sulfate) were purchased from Sigma. Ciprofloxacin hydrochloride was obtained from Sangon Biotech (Shanghai, China). DNA topoisomerase I (E. Coli) was purchased from Novoprotein (Shanghai, China).
Preparation of ciprofloxacin hydrazide: Ciprofloxacin hydrazide was synthesized based on a literature method (Hu et al., 2012). The ciprofloxacin hydrazide was characterized by IR (FTIR-21, Shimadzu, Japan) and 1H NMR ((Nuclear Magnetic Resonance) 400 MHz, Bruker) spectrum: vNH: 3400 cm-1 (broad, very strong); vCO (amide): 1700 cm-1 (medium); 1H NMR (d6-DMSO, ) δ: 1.18-1.32 (m, 4H, CH2CH2), 3.16-3.24 (m, 8H, piperazine-H), 3.35 (m, 1H, CH), 3.83 (s, 2H, NH2), 7.54 (d, J = 13.5 Hz, 1H, 5-H), 7.89 (d, J = 7.2 Hz, 1H, 8-H), 8.66 (s, 1H, 2-H), 10.57 (s, 1H, NH).
Bacteria: Staphylococcus aureus (ATCC(American Type Culture Collection) 43300), Fecal coliform (ATCC 29212), Staphylococcus epidermidis (ATCC 12228), Salmonella (ATCC 25923) and Saprophyte (ATCC 27853) were purchased from the American Type Culture Collection (gifts from Clinic Laboratory, Xinxiang Medical University). All the bacteria inocula for the well diffusion tests were derived from cultures that had been aerobically grown for 6 h in LB (lysogeny broth) medium (Difco) at 37°C.
The agar well diffusion tests were performed on LB agar plates. One freshly grown colony of the bacterial strain was inoculated into 2 mL of LB medium. The culture was then incubated as described above. The density of the inoculum was determined by OD 600 nm. The inoculum (2 mL) was mixed with a total of 50 mL of agar medium (~37°C) and then was poured into several petri dishes (150 by 15 mm) to a depth of ~4 mm. A series of wells were made by an autoclaved hole puncher in a laminar flow bench once the agar solidified. The 30 μL of 12.5, 25 and 50 μM ciprofloxacin (or it hydrazine derivative) was pipetted into the well. Inhibition zones developed due to active antimicrobial metabolites were measured after 24 h of incubation. The experiments were repeated 3 times.
Cytotoxicity assay: Bel-7402 and HepG2 human hepatoma cell lines (Y-S Biotechnology Inc., Shanghai, China) were grown in RPMI 1640 (Roswell Park Memorial Institute 1640) medium supplemented with 10% freshly inactivated Fetal Calf Serum (FCS) and 100 units mL-1 penicillin G and 100 μg mL-1 streptomycin in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The cells in exponential-phase were suspended in fresh RPMI-1640 and equal cells were seeded equally (5x103/well) into 96-well plate. When the cells attached in a humidified atmosphere of 5% CO2, the test compounds were added with final concentrations at 1000, 500, 250, 125, 60, 30 and 15 μM. After incubation for 48 h, MTT solution of an appropriate concentration (1 mg mL-1) was added to each well, the plate was further incubated for 4 h. The cell culture medium was removed by aspiration and 100 μL DMSO (Dimethyl Sulfoxide) was added into each well. The measurement of absorbance at 492 nm was performed on an ELISA (Enzyme-linked immunosorbent assay) spectrophotometer (MK3, Thermo Scientific). Percent growth inhibition was defined as percent absorbance inhibition within appropriate absorbance in each cell line. The same assay was done in triplet.
RT-PCR (Reverse transcription polymerase chain reaction ): Total RNA was extracted from the HepG2 cells that exposed to IC50 dose of the test compounds for 24 h using Trizol reagent (Shangong biological, Shanghai, China) according to the Manufacturer’s protocol. The 3 micrograms of total RNA were used for reverse transcription in a total volume of 20 μL following manufacturer’s recommendation (Lifefeng, Shanghai, China). Aliquots of 2 μL cDNA were subsequently amplified in a total volume of 25 μL. The sense and antisense primer for beta actin were 5’-ACACTGTGCCCATCTACGAGG-3’and 5’-CGGACTCGT CATACTCCTG CT- 3’(615 bp) that were used as an internal control; the sense and antisense primers for bcl2 were 5’-TTACCAAGCAGCCGAAGA-3’ and 5’-TCCCTCC TT TACATTCACAA -3’ (309 bp, NM_138621); the sense and antisense primers for bax, 5’- TTTTGCTTCA GGGTTTCA TC-3’ and 5’- GGCCTTGAGCACCAGT TT-3’(299 bp, BC014175); the sense and antisense primers for p53, 5’-GTCTACCTCCCGCCATAA-3’, 5’-CATCT CCCA AACAT CCCT-3’ (316 bp, M_ 001126114.2), respectively. The cycling conditions: 94°C for 5 min, followed by 28 cycles of 94°C for 30 sec, 58°C for 30 sec and 72°C for 1 min and a final extension of 72°C for 10 min. PCR products were separated on the 1.5% agarose gel viewed by ethidium bromide staining. These data were acquired with Tocan 360 gel imager (version 3.2.1 software). The RT-PCR experiments were done in duplicates.
Cell cycle analysis: HepG2 cells (1x105) were seeded in a six well plate. After 24 h incubation at 37°C (5% CO2), the medium was changed with fresh, supplemented or not (control) with the ciprofloxacin or the hydrazide (100, 200 μM). After 24 h of incubation, cells were harvested, washed with PBS, fixed in 70% ethanol and stored at -20°C for 1 h. Followed by removing the ethanol, washed with PBS, the cells were suspended in 0.5 mL PBS containing 50 μg mL-1 PI (propidium iodide) and 100 μg mL-1 RNase and incubated at 37°C for 30 min. Flow cytometry was performed in duplicate with a FACScalibur flow cytometer (Becton Dickinson, USA). Each sample 10000 events were collected and fluorescent signal intensity was recorded and analyzed by CellQuest and Modifit (Becton Dickinson, USA).
Flow cytometry analysis of mitochondrial membrane potential: Following similar procedure as mentioned in cell cycle analysis, the HepG2 cells were treated with ciprofloxacin or ciprofloxacin hydrazide (100 and 200 μM) for 24 h, the cells were washed with PBS twice after removing the cell medium and digested with trypsin. Treated cells were collected and resuspended at a concentration of 1x105 mL-1 in PBS containing 1 μmol L-1 Rhodamine123 and then incubated at 37°C for 30 min for directly using in flow cytometry. Samples were analyzed by FACSCalibur flow cytometer (Becton Dickinson, USA) with an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Data collection and analysis were as described above.
Microfluorimetry assessment of mitochondrial membrane permeability: The HepG2 cells were treated as did in flow cytometry analysis of mitochondrial membrane potential, but the cells were stained by 20 ng mL-1 of Rhodamine123 (R123) in PBS for 30 min. The slip was placed on the stage of the NikonFN1 microscopy equipped with EMCCD and visualized through a 40xwater-immersion objective. The intensifier gain on the camera was always set at the same value for consistency of fluorescent readings and allowing direct comparison between groups. The R123 excitation light (490 nm) was provided by a monochromator (Cairn Research, Ltd., UK). The emission filters (550-nm cutoff for R123) were placed in a Sutter filter wheel installed in front of the camera. Both excitation and emission conditions were controlled by the software (MetaFluor, Molecular Devices Corp., Downington, PA, USA). Images were captured using the MetaFluor/Meta-Morph software (Molecular Devices Corp.).
DNA topoisomerase activity assay: Crude nuclear extract from HepG2 cell line was prepared as described (Eijdems et al., 1995). Nuclear extract (0.4 μg) was added to the topoisomerase reaction mixture containing 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 150 mM NaCl, 0.1% BSA(Bovine serum albumin), 0.1 mM spermidine, 5% glycerol and 0.4 μg pUC18 and 3 μL (or 2, or 1 μL) test compound (1 mM) at a final volume of 20 μL. Following incubation at 37°C for 30 min, the reaction was terminated by adding 5 μL of stopping buffer (10% SDS, 0.025% bromophenol blue and 5% glycerol).
Similar procedure was used for inhibition of bacterial DNA topoisimerase I (Shanghzi China) except working in a total volume of 25 μL, 0.6 μg pUC18, 100~400 μM test compound and 1 unit DNA topoisomerase I included in reaction mixture. The reaction products were analyzed by electrophoresis on 1% agarose gel using a TBE buffer with 0.1% SDS (89 mM Tris-HCl, 89 mM boric acid and 62 mM EDTA) at 45 V for 3 h. The agarose gel stained by ethidium bromide (0.5 μg mL) and photographed on Tocan 360 gel scanner (Shanghai Tiancheng Technology Inc, China). The assay was conducted in duplicates.
Molecular docking studies: The structure of bacterial type II DNA topoisomerase with DNA and ciprofloxacin (2XCT) and human topoisomerase I (1K4T) were obtained from RCSB Protein Data Bank (Bax et al., 2010). The ciprofloxacin hydrazide was generated from Chemdraw (Chemdraw Ultra 8.0), the energy minimization was conducted by Chem3D (Ultra 8.0) (Ok et al., 2013). The resulting models were displayed in PyMol (The PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC).
Molecular docking studies were performed by AutoDock Vina and AutoDock Tools based on the recommended procedure (Trott and Olson, 2010). Grid boxes were set to the center of ciprofloxacin (or topotecan) model and the grid box size for ciprofloxacin hydrazide derivative models was set to 22, 24 and 28 for X, Y and Z axes, respectively. The ciprofloxacin hydrazide was set as a flexible ligand by using the default parameters of the AutoDock Tools. The optimal conformation of the ligand was generated by Autodock Vina.
Statistical analysis: Values shown are Means±S.D. for the indicated number of independent experiments. Statistical significance was assessed by Student t-test and expressed as follows: * p<0.05.
RESULTS
To investigate the substituent effect on the positions 3 of ciprofloxacin (Fig. 1), we transformed the 3-carboxylate to hydrazide by reaction of ciprofloxacin with hydrazine based on the reported method (Hu et al., 2012).
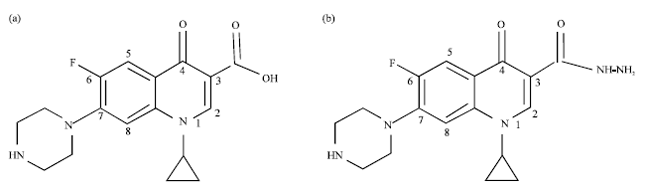 | |
| Fig. 1(a-b): | Chemical structures of (a) ciprofloxacin (cip) and (b) Its hydrazide derivative ciprofloxacin hydrazide (cip-D) |
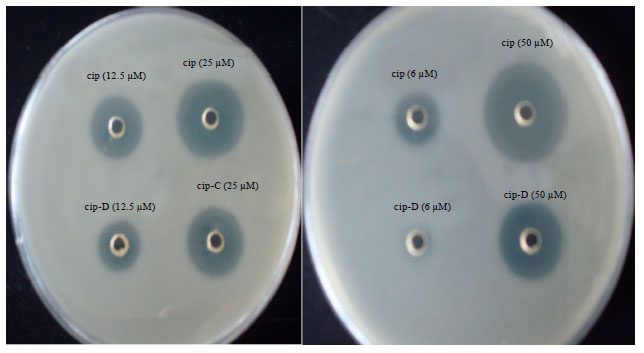 | |
| Fig. 2: | Well diffusion assay of ciprofloxacin and its hydrazide derivative against Staphylococcus aureus at indicated concentration |
The resulting hydrazide derivative of it was characterized by IR spectrum and 1H NMR. Compared to ciprofloxacin, a broad strong peak at 3400 cm-1 was appeared, which was assigned to N-H (Sahoo et al., 2012), while the peak at 3540 cm-1 (assigned to íOH) was disappeared, indicating the -OH of carboxyl was substituted with -NH-NH2 group. The 1H NMR was also supportive of the ciprofloxacin hydrazide formed.
Effect of substitution of carboxyl with hydrazide on antimicrobial activity: The structural modification may alter antibacterial spectra of the parent compound. To determine the effect on antibacterial activities, the ciprofloxacin hydrazide inhibitions of Staphylococcus aureus, Fecal coliform, Staphylococcus epidermidis, Salmonella and Saprophyte were evaluated using agar well diffusion tests, the diameters of the circular inhibition zones were measured after incubation at 37°C for 14 h, as shown in Fig. 2. The inhibition abilities of ciprofloxacin hydrazide and ciprofloxacin against the investigated bacterial strains are shown in Fig. 3. In terms of antibacterial activities, ciprofloxacin hydrazide was weaker than ciprofloxacin. It was interesting that the antimicrobial activities were bacterial strain dependent. The hydrazide derivative exhibited 70~85% inhibition efficiency compared to ciprofloxacin (as shown in Fig. 3), for Staphylococcus aureus, Fecal coliform, Staphylococcus epidermidis and Saprophyte. However, the difference was not significant for Samonella strain, but a large difference was observed at the minimum inhibitory concentration (6 μM) for Saprophyte strain (Fig. 3).
Inhibitory effect of ciprofloxacin and its derivative on HCC cell lines: To determine whether there was change in antitumor activity after the substitution of -OH of carboxyl to -NHNH2, MTT assay was employed to evaluate cytotoxicity of the hydrazide derivative against HepG2 and Bel-7402 cell lines (Fig. 4).
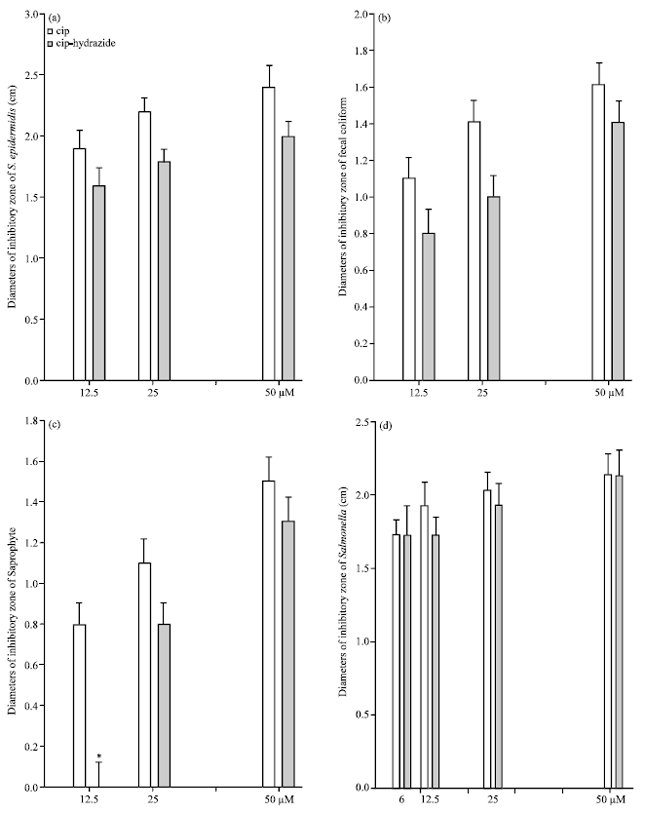 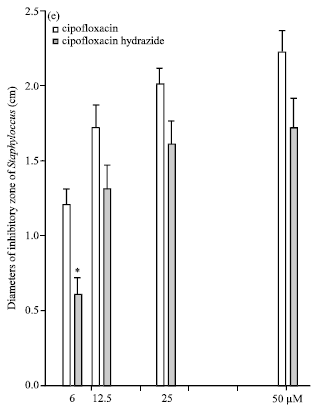 | |
| Fig. 3(a-e): | Comparison of antimicrobial activities of ciprofloxacin and ciprofloxacin hydrazide (a) S. epidermidis, (b) Fecal coliform, (c) Saprophyte, (d) Salmonella and (e) Staphylococcus aureus as indicated in each panel. Results represent Mean±SD for 3 independent experiments. * p<0.05 |
For Bel-7402 cell line, hydrazide derivative seemed to be more toxic than that of ciprofloxacin (IC50: 212±19 μM for ciprofloxacin, 172±12 μM for ciprofloxacin hydrazide), while for HepG2 cell line, a reversed results were shown (IC50: 284±12 μM for ciprofloxacin, 348±15 μM for ciprofloxacin hydrazide). The results indicated that the antitumor ability of hydrazide derivative was not altered after the structural modification compared to ciprofloxacin.
Effect of the structural change on gene expressions: The slight difference in antimicrobial and antitumor activity after the structural modification has been shown as mentioned above, it is necessary to determine whether change in action mechanism. Ciprofloxacin induces apoptosis via regulation of pro-apoptotic Bax, anti-apoptotic Bcl-2 genes to exert its cytotoxicity, so RT-PCR was employed to evaluate the gene regulation of the derivative. As shown in Fig. 5a, Bax increased dose-dependently after 24 h of incubation in the tested HepG2 cell line, while Bcl-2 remained unchanged during the observation time. For easy comparison, the ratio of Bax/Bcl-2 was set at 1.0 in untreated cells. As shown in Fig. 5b, the Bax/Bcl-2 ratio in the ciprofloxacin treated cells remained close to the control, whereas it significantly increased in the ciprofloxacin hydrazide treated group. There was no obvious change in p53 gene expression after the drug treatment at the given concentrations.
Ciprofloxacin hydrazide induces S phase arrest: To evaluate whether the structural modification affected cell cycle arrest, cell cycle analyses were conducted after 24 h exposure of hydrazide derivative or ciprofloxacin on HepG2 cells. As shown in Fig. 6, both drugs caused an accumulation of cells in the S-phase. The percentage of cells at the S-phase significantly increased from 21.56 to 24.65 and 37.31% after treatment with 100 and 200 μM ciprofloxacin, respectively. Similarly results were observed with ciprofloxacin hydrazide where the S phase accumulation was changed from 21.56 to 26.68 and 40.45% after treatment with 100 and 200 μM of the investigated drugs. A net ~3% increase in population in S phase arrest could be seen between them and their inducing ability was concentration dependent. These results indicated that ciprofloxacin and its derivative exert their anti-proliferative effect on HepG2 cells by inducing cell cycle arrest and the minor structural modification did not alter ciprofloxacin’s action mode on host cell.
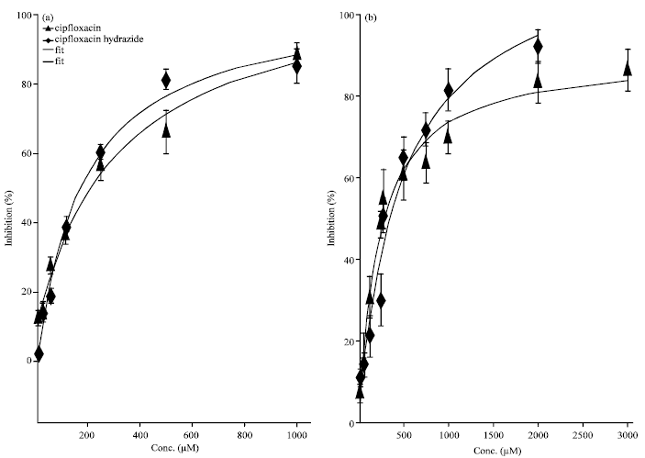 | |
| Fig. 4(a-b): | Comparison of antitumor activities of ciprofloxacin and ciprofloxacin hydrazide (a) Bel-7402 and (b) HepG2. The HCC cell lines are indicated in each panel. The MTT assays were done in triplicates. Results represent Mean±S.D |
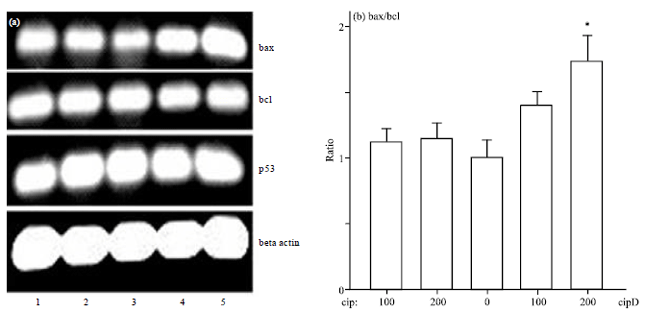 | |
| Fig. 5(a-b): | Gene regulation of ciprofloxacin (cip) and its hydrazide derivative (cip-D), (a) 1, 200 μM ciprofloxacin, 2, 100 μM ciprofloxacin, 3, control, 4, 100 μM ciprofloxacin hydrazide, 5, 200 μM ciprofloxacin hydrazide and (b) Normalized ratio of bax/bcl2 at 100 or 200 μM of ciprofloxacin or its derivative. The experiments were repeated in duplicates |
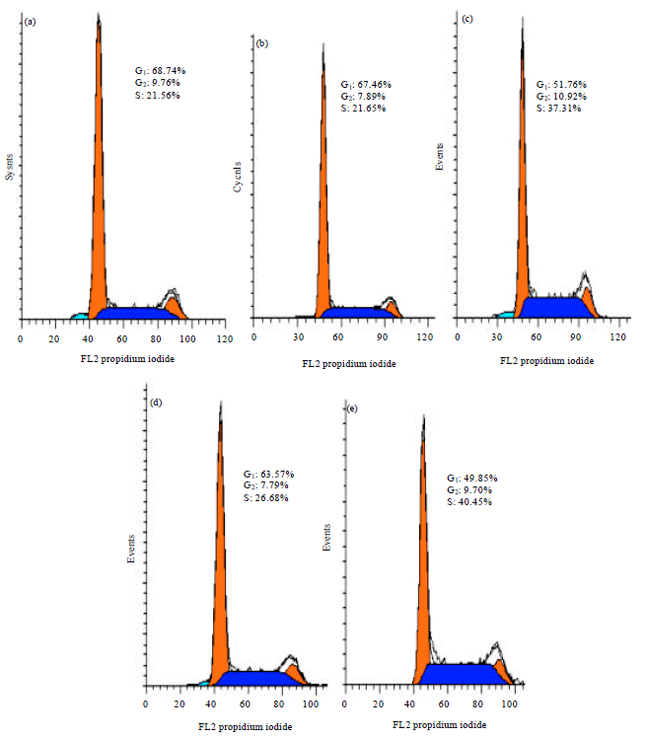 | |
| Fig. 6(a-e): | Effect on HepG2 cell cycle, (a) Control, (b) 100 μM ciprofloxacin, (c) 200 μM ciprofloxacin, (d) 100 μM ciprofloxacin hydrazide and (e) 200 μM ciprofloxacin hydrazide |
Mitochondrial membrane potential: Ciprofloxacin can depolarize the mitochondrial membrane and inhibit mtDNA synthesis, so it is necessary to determine whether there was change after the structural modification. The depolarization of mitochondrial membrane was evaluated via rhodamine123 staining on BD flow cytometry and microfluorimetry. The results are shown in Fig. 7. Compared to control, the distribution of cells with different fluorescence and cellular sizes was altered in both ciprofloxacin and its derivative groups. A 5.9% increase in M2 gate between the drugs at 100 μM the drugs, while 6.3% decrease at 200 μM could be seen (Fig. 7). For the purpose of intuitive comparison, the photo-images were taken on microfluorimetry, the images are shown in Fig. 8. The trend in cellular fluorescence after the drug treatment was consistent with the results from flow cytometry.
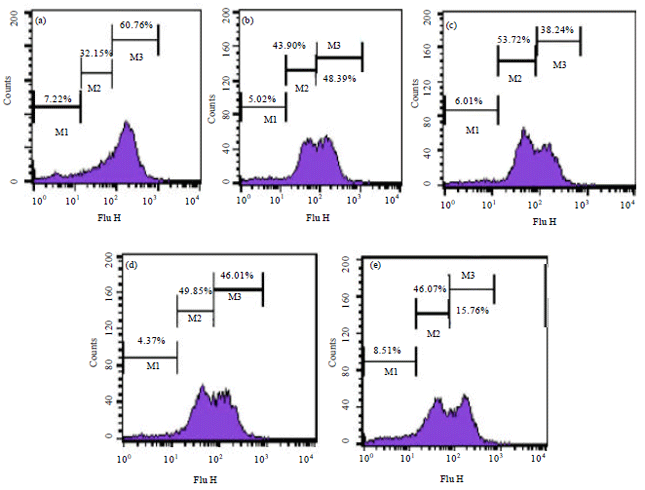 | |
| Fig. 7(a-e): | Effect on HepG2 cell mitochondrial membrane potential, (a) Control, (b) 100 μM ciprofloxacin, (c) 200 μM ciprofloxacin, (d) 100 μM ciprofloxacin hydrazide and (e) 200 μM ciprofloxacin hydrazide. A 5.9% increase in M2 gate between b and d; 6.3% decrease in M2 between c and e, which shown the difference between ciprofloxacin and its derivative in mitochondrial depolarization |
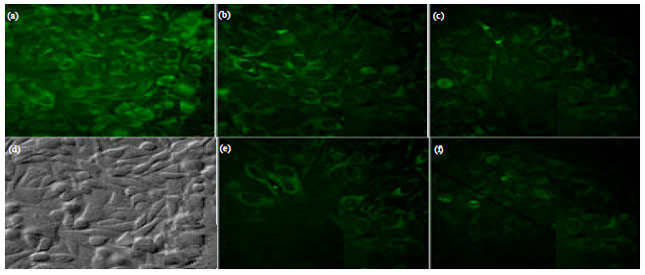 | |
| Fig. 8(a-f): | Effect of ciprofloxacin and its hydrazide derivative on mitochondrial membrane permeability, (a) Control, (b) 100 μM ciprofloxacin, (c) 200 μM ciprofloxacin, (d) Control 100 μM ciprofloxacin hydrazide, (e) 100 μM ciprofloxacin hydrazide and (f) 200 μM ciprofloxacin hydrazide |
 | |
| Fig. 9(a-b): | Ciprofloxacin (cip) and ciprofloxacin hydrazide (cip-D) inhibition of (a) Human and (b) Prokaryote DNA topoisomerase. Panel A: 1: 300 μM ciprofloxacin hydrazide, 2: 200 μM ciprofloxacin hydrazide, 3: 100 μM ciprofloxacin hydrazide, 4: ciprofloxacin hydrazide plus pUC18, 5: Markers, 6: pUC18 plus nuclear extract, 7: pUC18, 8: ciprofloxacin plus pUC18, 9: 300 μM ciprofloxacin, 10: 200 μM ciprofloxacin, 11: 100 μM ciprofloxacin. Panel B: 1, 400 μM ciprofloxacin, 2: 200 μM ciprofloxacin, 3: 100 μM ciprofloxacin, 4: pUC18, ciprofloxacin hydrazide plus pUC18, 5: pUC18 plus type I topoisomerase, 6: 100 μM ciprofloxacin hydrazide, 7: 200 μM ciprofloxacin hydrazide, 8: 400 μM ciprofloxacin hydrazide |
These observations indicated that both ciprofloxacin and its hydrazide derivative could depolarize the mitochondrial membrane, decreasing its electrochemical potential.
Effect of the structural change on inhibition of prokaryotic DNA topoisomerase and human topoisomerase I: The ciprofloxacin as type II DNA topoisomerase inhibitor was well documented. To investigate whether the molecular mechanism in antimicrobial and antitumor activities was altered after the minor change in structure, the inhibition of human topoisomerase by ciprofloxacin hydrazide was conducted using nucleic extract. As shown in Fig. 9a, the ciprofloxacin derivative exhibited inhibition of human topoisomerase as did ciprofloxacin, its inhibitory activity seemed to be the same or a little stronger than ciprofloxacin. Similarly the inhibitory assay of the drugs against prokaryotic topoisomerase I was also evaluated (Fig. 9b), the result indicated that there is no obvious change after the minor modification in structure. Therefore, we speculated that the molecular basis for antimicrobial and antitumor characteristic was not altered by the structural modification.
Molecular docking analysis: The experimental evidences demonstrated that the substitution of -OH by -NH-NH2 on position 3 of ciprofloxacin did not significantly change the biological activities of these drugs, but minor differences could be seen, especially in antimicrobial activity. To understand the structural basis for the phenomenon the molecular docking was performed. Autodock is a widely used molecular docking software. Based on the recommended procedure, the ciprofloxacin-free type II topoisomerase derived from 2XCT (PDB ID: 2XCT) was chosen as receptor and the ciprofloxacin hydrazide or ciprofloxacin was used as ligand. The input pbdqt files were generated by AutoDockTool and the optimal conformations of ligand were calculated on Autodock Vina. The calculated data indicated that the docked ciprofloxacin hydrazide has an -9.3 kcal mol-1 affinity energy with the DNA-topoisomerase complex. For easy comparison, the ciprofloxacin was re-docked in the crystal structure, resulting a -9.6 kcal mol-1 affinity energy with the complex, showing a slight difference in binding affinity between the ligands. The orientations of the ligands were shown in Fig. 10a, b. Similarly the same procedure was used for docking the ligands into human type I topoisomerase, showing the binding affinity with a -8.5 kcal mol-1 for ciprofloxacin hydrazide, -8.2 kcal mol-1 for ciprofloxacin, respectively. Compared to topotecan, an anticancer drug in clinical treatments (-12.7 kcal mol-1, Fig. 11), the interaction of the ligands with the type I topoisomerase was much weaker. Interestingly, an almost opposite orientation can be seen even though the ligands have similar binding affinity.
DISCUSSION
Ciprofloxacin remains as one of the safest of all antibiotics (Ball et al., 1999), so the ciprofloxacin based drug design is still an interesting research topic (Bartzatt et al., 2010).
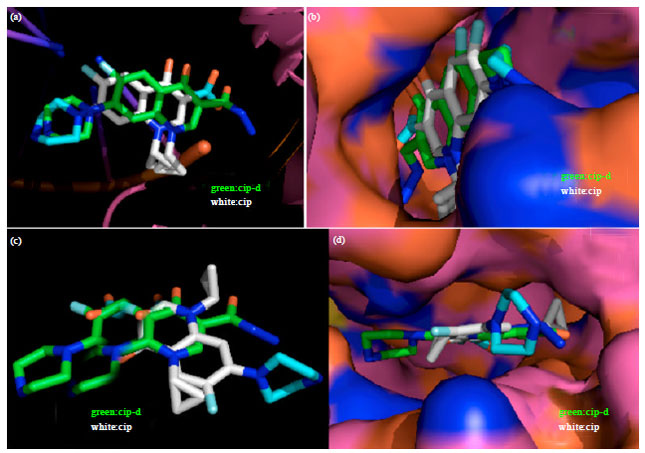 | |
| Fig. 10: | Docking of two inhibitors, ciprofloxacin (cip) and ciprofloxacin hydrazide (cip-d), into the active site of prokaryotic type II topoisomerase (PDB ID: 2XCT) and human type I topoisomerase (PDB ID: 1K4T). (a) Orientation of inhibitors at DNA cleavage site of topoisomerase of E. coli, (b) Inhibitors shown on the DNA cleavage cavity of the prokaryotic topoisomerase, (c) Orientation of inhibitors at DNA cleavage site of human topoisomerase I and (d) Inhibitors on the binding pocket of the human type I topoisomerase |
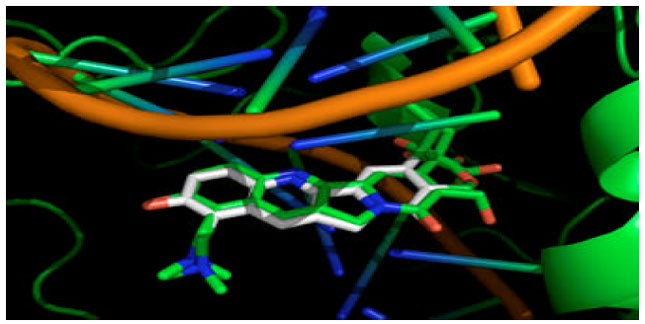 | |
| Fig. 11: | Docking of topotecan on human type I topoisomerase (PDB ID: 1K4T). The orientations of topotecan docked (green) and in crystal structure (white) are shown in the figure |
It has been shown that the carboxylate group on the position 3 of ciprofloxacin is essential for antimicrobial activity. The crystal structure of ciprofloxacin complex with Staphylococcus aureus DNA gyrase and DNA reveals that ciprofloxacin is close to the active sites. It could imagine that the larger substitution on position 3 of ciprofloxacin may alter (move away) significantly the interaction between the gyrase and ciprofloxacin and compared with carboxyl group the amide (-CONH2) will not change remarkably in view of steric structure, but the hydrazide substituent (NHNH2) seems to move the ciprofloxacin away from the binding site. If it was true, the substitution will eliminate the antimicrobial activity of ciprofloxacin. To test the hypothesis, the ciprofloxacin hydrazide derivative was made to testify the effect of one chemical bond longer on the biological property. Our results demonstrated that the substitution the -OH of carboxylate group to hydrazide on the position 3 of ciprofloxacin did not significantly affect its antimicrobial, but the slight difference (weaker in antimicrobial activity) after the structural modification can be seen. It may demonstrate that the gyrase in solution is more flexible to accommodate the derivative than in solid. In addition to antimicrobial activity, ciprofloxacin has a certain degree of anti-tumor activity (Bourikas et al., 2009), so could its hydrazide derivative. Our group and others have determined the proliferation inhibition of ciprofloxacin against hepatic cancer cells (Kloskowski et al., 2011; Fu et al., 2013), so the cell lines were used for evaluation of the new ciprofloxacin derivative. Our data clearly showed that there are differences in the apoptotic gene regulation between ciprofloxacin and its derivative the Bax/Bcl-2 ratio in the ciprofloxacin treated HepG2 cells was close to the control, but the ratio increased significantly in the ciprofloxacin hydrazide treated cells (Fig. 5). This indicates both ciprofloxacin and its hydrazide derivative induce apoptosis via the same molecular path. The cell cycle analysis indicated that ciprofloxacin and its hydrazide derivative have similar ability to move more cells into S phase.
Mitochondria produce ATP by utilizing the proton electrochemical gradient potential. The depolarization of mitochondrial membrane will result in decreases of the potential and proton gradient between inner and outer mitochondrial membrane, consequently lead to ATP production lesser efficient (Perry et al., 2011). Our observations also supported that both ciprofloxacin and its hydrazide have the function of depolarization of mitochondrial membrane compared to the untreated group from the fluorescence intensity of mitochondria stained by rhodamine123, which may slow down cell growth. In comparison with ciprofloxacin, the effect of ciprofloxacin hydrazide on permeability of mitochondrial membrane seemed to be more evidence from both in flow cytometry and microscopy experiments. The effect of ciprofloxacin may be due to unselective inhibition of mitochondrial DNA-synthesis with subsequent mitochondrial injury, so it is reasonable to speculate that the disturbance of mitochondrial respiration and ATP synthesis may contribute to the cytotoxic effect of ciprofloxacin or its hydrazide derivative which is in a concentration-dependent manner (Rawi et al., 2011; Koziel et al., 2006).
Quinolones induce cell death by causing double-stranded DNA breakage and inhibiting topoisomerase function (Drlica et al., 2008). Both prokaryotic and human topoisomerase inhibition by ciprofloxacin and its hydrazide at higher concentration were also observed in our experiments (Fig. 9), which may reduce synthesis of mitochondrial DNA. Meanwhile, energy depletion promotes apoptosis via induction of cell cycle arrest (Bratton et al., 2000). The fact that both ciprofloxacin and its hydrazide can induce cells to the S phase is consistent with that reported in the literature. The inhibitory ability of the hydrazide derivative for bacterial topoisomer I was similar to ciprofloxacin, but seemed to be stronger in human topoisomerase inhibition (Fig. 9). The weaker inhibition (poor anticancer activity) of human topoisomerase by ciprofloxacin and its hydrazide derivative was consistent with that reported in literature (Drlica, 1999).
Molecular docking is a well established computational technique to predict the interaction between enzyme and inhibitor (Huang, 2009; Glaser et al., 2006; Fukunishi and Nakamura, 2011). Autodock was used to predict the binding information of test compounds (Takatsuka et al., 2010; Nikaido and Pages, 2012). Compared to ciprofloxacin at the binding pocket, the orientation of the hydrazide derivative was similar to its parent compound and its affinity to the topoisomerase was also decreased slightly. This was constant with the results in antimicrobial activity for the subtle changes were observed when exposure to ciprofloxacin and its derivative. To eukaryotic topoisomerase, the docking results indicated that the affinity of ciprofloxacin hydrazide was slight increased, but their binding affinity was quite weaker compared to topotecan. These might be correlated to their poor antitumor activity.
In summary, although the structural modification on position 3 of ciprofloxacin with the -NH-NH2 group could alter slightly antimicrobial spectra of ciprofloxacin in dose and strain dependent manner, overall it was not significant. Likewise, there was no obvious difference in antitumor activities. Mechanistic studies revealed that the hydrazide derivative exhibited inhibition of topoisomerase, depolarization of mitochondrial membrane, pro-apoptotic gene regulation and cell cycle arrest, which were similar to ciprofloxacin, indicating that the structural modification did not alter the molecular targets.
ACKNOWLEDGMENT
We thank Dr. Huiying Li for critical reading of the manuscript (Department of Molecular Biology and Biochemistry, University of California, Irvine), Dr. Yuting Yang for help in molecular docking experiment (Department of Anesthesia, University of Michigan), Ms. Xia Wang for help in flow cytometry experiments (Department of Laboratory, Xinxiang Medical University). This work was supported by Grants CP1204 from Xinxiang Scientific and Technology Division; ZD2011-06 from Xinxiang Medical University.
REFERENCES
- Ahmed, A. and M. Daneshtalab, 2012. Nonclassical biological activities of quinolone derivatives. J. Pharm. Pharm. Sci., 15: 52-72.
PubMed - Ball, P., L. Mandell, Y. Niki and G. Tillotson, 1999. Comparative tolerability of the newer fluoroquinolone antibacterials. Drug Saf., 21: 407-421.
CrossRefDirect Link - Bourikas, L.A., G. Kolios, V. Valatas, G. Notas and I. Drygiannakis et al., 2009. Ciprofloxacin decreases survival in HT-29 cells via the induction of TGF-b1 secretion and enhances the anti-proliferative effect of 5-fluorouracil. Br. J. Pharm., 157: 362-370.
PubMed - Bratton, S.B., M. MacFarlane, K. Cain and G.M. Cohen, 2000. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp. Cell. Res., 256: 27-33.
CrossRefDirect Link - Drlica, K., M. Malik, R.J. Kerns and X. Zhao, 2008. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother., 52: 385-392.
CrossRefPubMedDirect Link - Eijdems, E., M. Haas, A.J. Timmerman, G.P.V. Schans and E. Kamst et al., 1995. Reduced topoisomerase II activity in multidrug-resistant human non-small cell lung cancer cell lines. Br. J. Cancer, 71: 40-47.
Direct Link - Fabian, I., D. Reuveni, A. Levitov, D. Halperin, E. Priel and I. Shalit, 2006. Moxifloxacin enhances antiproliferative and apoptotic effects of etoposide but inhibits its proinflammatory effects in THP-1 and Jurkat cells. Br. J. Cancer, 95: 1038-1046.
CrossRefPubMedDirect Link - Fukunishi, Y. and H. Nakamura, 2011. Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library. Protein Sci., 20: 95-106.
PubMed - Fu, Y., S. Zhou, D. Li, Y. Zhang, S. Li and C. Li, 2013. Ciprofloxacin inhibits proliferation and synergistic effect against hepatocellular carcinoma cancer lines with cisplatin. Afr. J. Pharm. Pharmacol., 7: 1793-1801.
CrossRefDirect Link - Glaser, F., R.J. Morris, R.J. Najmanovich, R.A. Laskowski and J.M. Thornton, 2006. A method for localizing ligand binding pockets in protein structures. Struct. Funct. Bioinf., 62: 479-488.
CrossRefDirect Link - Hawtin, R.E., D.E. Stockett, J.A.W. Byl, R.S. McDowell and N. Tan et al., 2010. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS ONE, Vol. 5.
CrossRef - Hu, G.Q., Y.S. Chen, G.Q. Wand, N.N. Duan and X.Y. Wen et al., 2012. Part IV. Synthesis and antitumor evaluation of s-triazolothiadiazines and pyrazolo s-triazoles derived from ciprofloxacin. Acta Pharmaceut. Sin., 47: 66-71.
PubMed - Jubie, S., P. Sikdar, R. Kalirajan, B. Gowramma, S. Gomathy and S.K. Sankar, 2010. Synthesis and antimicrobial activity of some novel ciprofloxacin analogues. J. Pharm. Res., 3: 511-513.
Direct Link - Kloskowski, T., N. Gurtowska, M. Nowak, R. Joachimiak, A. Bajek, J. Olkowska and T. Drewa, 2011. The influence of ciprofloxacin on viability of A549, HepG2, A375.S2, B16 and C6 cell lines in vitro. Acta Pol. Pharm., 68: 859-865.
PubMedDirect Link - Koziel, R., K. Zablocki and J. Duszynski, 2006. Calcium signals are affected by ciprofloxacin as a consequence of reduction of mitochondrial DNA content in Jurkat cells. Antimicrob. Agents Chemother., 50: 1664-1671.
PubMed - Lemaire, S., F.V. Bambeke and P.M. Tulkens, 2011. Activity of finafloxacin, a novel fluoroquinolone with increased activity at acid pH, towards extracellular and intracellular Staphylococcus aureus, Listeria monocytogenes and Legionella pneumophila. Int. J. Antimicrob. Ag., 38: 52-59.
PubMed - Ok, K., Y.W. Jung, J.G. Jee and Y. Byun, 2013. Facile docking and scoring studies of carborane ligands with estrogen receptor. Bull. Korean Chem. Soc., 34: 1051-1054.
CrossRefDirect Link - Patitungkho, S., S. Adsule, P. Dandawate, S. Padhye, A. Ahmad and F.H. Sarkar, 2011. Synthesis, characterization and anti-tumor activity of moxifloxacin-copper complexes against breast cancer cell lines. Bioorg. Med. Chem. Lett., 21: 1802-1806.
PubMed - Rawi, S.M., I.M. Mourad, N.M.S. Arafa and N.I. Alazabi, 2011. Effect of ciprofloxacin and levofloxacin on some oxidative stress parameters in brain regions of male albino rats. Afr. J. Pharm. Pharmacol., 5: 1888-1897.
Direct Link - Sahoo, S., C.C. Kanti and P.K. Behera, 2012. Spectroscopic investigations of a ciprofloxacin/HPMC mucoadhesive suspension. Int. J. Appl. Pharm., Vol. 4.
Direct Link - Seo, K.W., R. Holt, Y.S. Jung, C.O. Rodriguez Jr., X.B. Chen and R.B. Rebhun, 2012. Fluoroquinolone-mediated inhibition of cell growth, S-G2/M cell cycle arrest and apoptosis in canine osteosarcoma cell lines. PLoS ONE, Vol. 7.
CrossRefDirect Link - Thadepalli, H., F. Salem, S.K. Chuah and S. Gollapudi, 2005. Antitumor activity of trovafloxacin in an animal model. In vivo, 19: 269-276.
PubMed - Tillotson, G.S., 1996. Quinolones: Structure-activity relationships and future predictions. J. Med. Microbiol., 44: 320-324.
PubMed - Trott, O. and A.J. Olson, 2010. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem., 31: 455-461.
CrossRefPubMedDirect Link - Yadav, P. and Y.C. Joshi, 2008. Syntheises and spectral studies of novel ciprofloxacin derivatives. Bull. Chem. Soc. Ethiop., 22: 459-464.
Direct Link - Devnath, H.P. and M.R. Islam, 2010. Synthesis of some pyrazolone derivatives from ciprofloxacin and study of their cytotoxicity. Bangladesh J. Pharmacol., 5: 30-33.
CrossRefDirect Link