
D. A. Al-Turki
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia
L. A. Abou-Zeid
Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, University of Mansoura, Mansoura 35516, Egypt
I. A. Shehata
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia
M. A. Al-Omar
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia
International Journal of Pharmacology
Year: 2010 | Volume: 6 | Issue: 6 | Page No.: 813-825







ABSTRACT
The discovery of the existence of two cyclooxygenases greatly refines the understanding of how the therapeutic and toxic effect of NSAIDs relate to inhibition of PG synthesis. As was reported, NSAIDs inhibit both COX-1 and COX-2 to different extents. This accounts for their anti-inflammatory and analgesic activities and also their unwanted GI side effect. Current evidence indicates that selective COX-2 inhibitors have important adverse cardiovascular effects that include increased risk for myocardial infarction, stroke, heart failure and hypertension. Thus, the development of selective COX-2 inhibitors could be a big step ahead in the therapeutic treatment of anti-inflammatory diseases with fewer risks and side effects. The selective COX-2 diaryl heterocyclic, Coxib group; is characterized by having different 1,2-diaryl five-membered or six-membered heterocycles that are considered as pharmacophore templates, such as, Celcoxib, Refecoxib, Valdecoxib and Etoricoxib. Moreover, the SAR studies have shown that the substituted sulfonyl group present in the structure of Coxibs, is considered one of the pharmacophoric moieties responsible for the selective recognition with the key amino acid residues at COX-2 active site pocket. Also, it has been reported that compounds having aryl methylsufone or aryl sulfonamide moieties display a propensity for COX-2 selectivity. Furthermore, taking in consideration the main difference between the two COX active sites which is the replacement of Ile523 in COX-1 by the less bulky Val523 in COX-2 active site, results in opening a polar side pocket that enlarges the volume of COX-2 active site and is considered a prerequisite for COX-2 drug selectivity.
PDF Abstract XML References Citation
Received: May 25, 2010;
Accepted: July 01, 2010;
Published: October 04, 2010
How to cite this article
D. A. Al-Turki, L. A. Abou-Zeid, I. A. Shehata and M. A. Al-Omar, 2010. Therapeutic and Toxic Effects of New NSAIDs and Related Compounds: A Review and Prospective Study. International Journal of Pharmacology, 6: 813-825.
DOI: 10.3923/ijp.2010.813.825
URL: https://scialert.net/abstract/?doi=ijp.2010.813.825
DOI: 10.3923/ijp.2010.813.825
URL: https://scialert.net/abstract/?doi=ijp.2010.813.825
INTRODUCTION
Inflammation is a normal and essential response to any noxious stimulus. The typical symptoms of inflammation are redness, swelling, local heat and the patient may be febrile. At the microscopic level, dilatation of the small blood vessels lead to increase in vascular permeability that leads to the leak of fluid and elements from blood into tissue spaces, leukocytes and other phagocytic cells migrate into the area and rupture of cell lysosomes releases lytic enzymes into the tissues. This process is accompanied by the local liberation of chemical mediators that include histamine, bradykinin and prostaglandins (Meyerson and Linderoth, 2006).
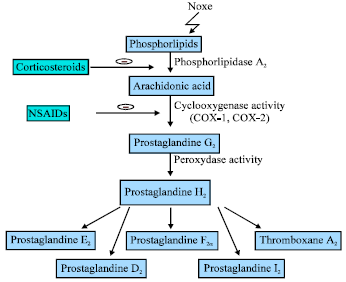
Prostaglandins (PGs) comprise a group of naturally occurring 20-carbon cyclopentano-fatty acid derivatives. They belong to a class of autacoid called eicosanoids derived from membrane phospholipids. Upon tissue exposure to any of the inflammation-precipitating factors, cell membranes release Arachidonic Acid (AA) by partial hydrolysis of lipids mediated by the membrane-bound enzyme phospholipase (Kalgutkar et al., 2000a). AA is subjected to one of two biochemical transformation routes. One route involves hydroxylation of the fatty acid by the enzyme lipooxygenase, resulting in the formation of a group of autacoids Called Leukotrines (LT). The second route involves oxygenation and a process of cyclization by cyclooxygenase enzyme (COX) to produce different types of PGs (Al-Turki, 2010; Vane and Botting, 1992; Flower and Vane, 1972; Vane, 1971; Wu et al., 2003) (Fig. 1).
Pgs are produced by most cells and also present in tissues, this explain their broad spectrum of biological responses. The outstanding effects of the PGs include their cycloprotective properties in the gastrointestinal (GI) tract and control renal functions in the kidney (Meyerson and Linderoth, 2006). The general structure of PGs is shown in Fig. 2.
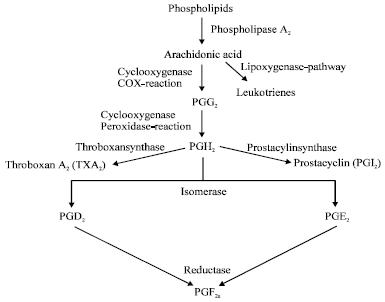 | |
| Fig. 1: | Arachidonic acid cascade (Al-Turki, 2010) |
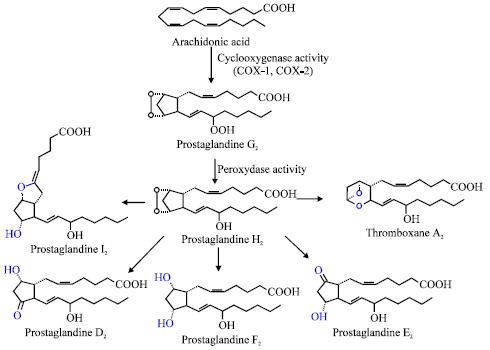 | |
| Fig. 2: | Chemical structure of prostaglandins (Al-Turki, 2010) |
The major effect of prostacycline (PGI2) is inhibition of the platelet aggregation process, while thromboxane (TXA2) has the opposite effect on platelets (Wu et al., 2003). Due to the apparent role of PGs in the process of inflammation, inhibiting PG biosynthesis has become an attractive approach to fighting inflammation.
ANTI-INFLAMMATORY DRUGS
Anti-inflammatory drugs; are type of drugs that influence the inflammatory process or its manifestations and they do so by a variety of actions. They include: Corticosteroids, immunosuppressive agents, colchicines, chloroquine, penicillamine, gold salt and non steroidal anti-inflammatory drugs (NSAIDs) of which Aspirin is the protype. Corticosteroids diminish inflammation of all types by preventing prostaglandin synthesis through inhibition of phospholipase A2 that releases the AA required. Long term use of corticosteroids poses many problems and in general this group of drugs should not be stopped immediately, gradual withdrawing under physician supervision is required (Kalgutkar et al., 2000b). NSAIDs will be the topic of concern in the following part.
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (NSAIDS)
Traditional NSAIDs are one of the most commonly used class of medication worldwide, primarily because of their effectiveness as anti-inflammatory, analgesic and antipyretic agents (Montvale, 2000). The first NSAID, sodium salicylate, was discovered in 1763, however, the side effects associated with the use of salicylates, particularly the GI toxicity, led to the introduction of non salicylate NSAIDs (Houston and Tech, 2004) (Fig. 3).
All drugs in this class cause adverse effects in a significant number of patients who use them and these are frequently dose limiting. None of the currently available NSAIDs is free of GI complications (Rodriguez et al., 1998).
Among the most widely prescribed drugs for treatment of rheumatic disorders and other degenerative inflammatory joint diseases are: Diclofenac (1), Indomethacin (2), Sulindac (3), Ketoprofen (4), Flurbiprofen (5), Ibuprofen (6), Naproxen (7), piroxicam (8), Tenoxicam (9), Tolmetin (10), Ketorolac (11) (Fig. 4).
Cyclooxygenase (COX Enzyme): The common mechanism of action for NSAIDs is inhibition of the synthesis of PGs by inhibiting the key regulatory COX enzyme (Fig. 5).
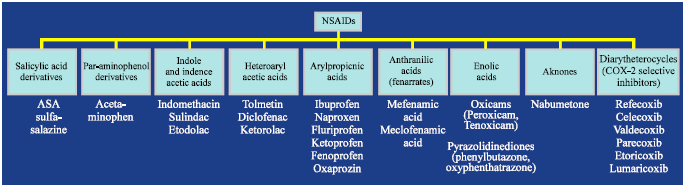 | |
| Fig. 3: | NSAIDs. The 9 chemical groupings of NSAIDs are shown, along with key compounds in each class |
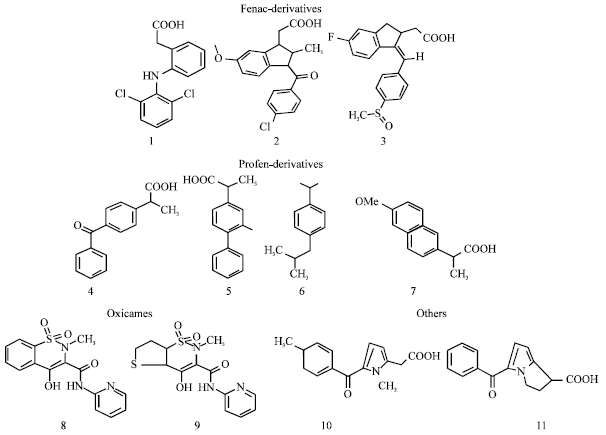 | |
| Fig. 4: | Classic NSAIDs |
 | |
| Fig. 5: | Mode of action of COX inhibitors |
In 1989, it was determined that there were at least 2 isoforms of cyclooxygenase: COX-1, or prostaglandin H1 synthase and COX-2, prostaglandin H2 synthase. COX-1 is expressed in most tissues, regulates physiologic processes such as gastric cytoprotection, kidney function and platelet aggregation and is stimulated by growth factors and hormones. It has been called the housekeeping enzyme (Sperling, 1995; Paloucek and Rynn, 2001; Lee, 2003; Singh, 1998; Graumlich, 2001; Simon, 2001; Noble et al., 2000).
Many toxic effects of NSAIDs, such as prolonged bleeding times and gastrointestinal side effects are attributed to the inhibition of COX-1 (Griffin et al., 1991; Hollander, 1994; Laine, 1996; Scheiman, 1996). COX-2 is found at low or undetectable levels in most tissues. It is an inducible enzyme whose expression is increased in response to inflammation or experimentally in response to mitogenic stimuli (Crofford et al., 2000; Dubois et al., 1998; Smith, 1992). COX-2 is constitutively expressed in the brain, specifically in the cortex, in the female reproductive system where it is associated with ovulation and implantation, in the male reproductive system, in bones where it is associated with osteoblast activity and in the kidney (Crofford et al., 2000; Dubois et al., 1998). In persons with normal kidney function, COX-2 facilitates the regulation of water and electrolyte balance (Chrischilles and Wallace, 1993; Handel and Nielsen, 1997; Johnson et al., 1993; Pope et al., 1993; Whelton, 1999).
Recently, COX-2 over expression has been demonstrated in several types of cancer, in angionenesis and in neurodegenerative diseases such as Alzheimer's or Parkinson's (Dannenberg et al., 2001; Shiff et al., 2003; Leahy et al., 2000; Masferrer et al., 2000; Hoozemans et al., 2003; Teismann et al., 2003).
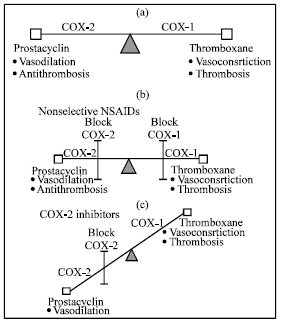 | |
| Fig. 6: | (a-c) The effects of COX-1 and COX-2 under normal conditions |
Prostaglandins produced by COX-2 are responsible for pain and inflammation where as those from COX-1 have a protective effect on the stomach lining, for this reason NSAIDs which blocks both COX-1 and COX-2 may cause peptic ulcer, while this unwanted effect is eliminated when COX-2 specific inhibitors are used.
Selective COX-2 inhibitors: Cyclooxygenase-2-inhibitors are a relatively new group of NSAIDs which at recommended doses block prostaglandin production by COX-2. Typical COX-2 inhibitors are drugs that show in vitro a minimum of 10-100 times stronger inhibition of COX-2 than COX-1 (Dewitt and Smith, 1988).
However, emerging evidence suggests that adverse reactions such as GI irritation or ulceration and renal liabilities are associated with prolonged use of COX-2 selective inhibitors (Kawaguchi et al., 1995). COX-2 selective inhibitors are also known to suppress synthesis of prostacyclin, a potent vasodilator, gastro-protectant and platelet inhibitors, via inhibition of COX-2. However COX-2 inhibitors do not inhibit production of thromboxane, a vasoconstrictor and promotor of platelet aggregation, which is synthesized in platelets by COX-1 (Silverstein et al., 1995; MacDonald et al., 1997) (Fig. 6a-c). Therefore, COX-2 inhibitors inttrinsically lack antithrombotic activity and some cardiovascular liabilities have been associated preclinically with them (Fries, 1991).
It has been reported that, COX-2 inhibitors have not been approved for use in children younger than 18 years old. In adults, candidate criteria for the use of COX-2 inhibitors over other NSAIDs have been suggested by Simon (2001): Age >60 years, History of GI bleeding, history of NSAID-induced GI toxicity, History of cardiovascular disease, requiring high-dose NSAIDs, concomitant use of glucocorticoids and requiring a combination of NSAIDs.
More than 500 COX-2 inhibitors have been described over the past few years. The large number of newly developed COX-2 inhibitors demonstrates how promising this group of anti-inflammatory agents is expected (Talley, 1999).
Classes of COX-2 inhibitors: From a structural point of view selective COX-2 inhibitors are divided into five classes (Gauthier et al., 2006): diaryl or aryl-heteroarylethers, structurally modified NSAIDs, compounds with antioxidative moieties, diarylethylene derivatives and vicinal diarylcarbocycles or heterocycles (Coxibs).
Diaryl or aryl-heteroarylethers: The first selective COX-2 inhibitor discovered in this class was compound NS-398 (12). It showed inhibition of PG synthesis in inflammatory cells and was free of unwanted GI effects in animal models.
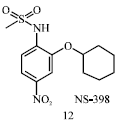 |
Nimesulide (13) and Flosulide (14) are tow more closely related members of this group of compounds that is characterized by having a methansulfonanilide moiety (Patrignani et al., 1994; Huff et al., 1995; Davis and Brogden, 1994; Li et al., 1995).
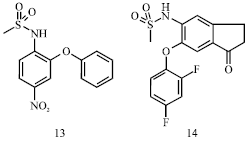 |
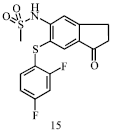
The thioether analogue of Flosulide L-745337 (15) was reported to have higher COX-2 specificity, better bioavailability, improved in vivo potency and greater GI safety than Flosulide.
 |
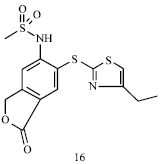
Furthermore a series of isobenzofuran derivatives was synthesized and compound 16 was reported to be the most potent member of this series in cell culture with COX-1: IC50 >100 μmol and COX-2: IC50 = 0.005 μmol.
 |
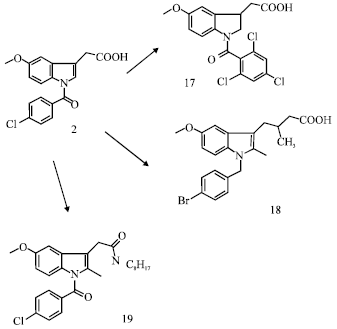
Structurally modified NSAIDs: Modifying well known NSAIDs into selective COX-2 inhibitors represents an interesting strategy (Dannhardt and Laufer, 2000). Classic NSAIDs such as indomethacin (2) possess both COX-1 and COX-2 inhibitory activity. Introduction of larger substituents as trichlorobenzoyl moiety and altering the side chain by a beta-branched butyric acid afforded compounds L-748780 (17) and L-761066 (18), respectively with high potency and remarkable activity (Black et al.,1996; Therien et al., 1996). However, it was reported that esterification or amide formation of the arylacetic acid moiety of indomethacin gave compound 19 capable of binding tightly to COX-2 but not to COX-1 (Kalgutkar et al., 2000b) (Fig. 7).
A similar strategy was used for modification of Zompirac (20) (Dannhardt and Kiefer, 2001; Saari and King, 1973), Flubiprofen (5) and Aspirin (21) (Kalgutkar et al., 1998) to obtain selective COX-2 inhibitors as shown in Fig. 8.
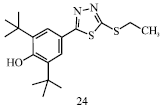
Compounds with antioxidative moieties: Song et al. (1999) reported that since COX enzyme catalysis involves radical intermediates, a radical scavenging moiety such as a di-tert-butylphenol interferes with COX reaction. Accordingly a series of compounds carrying this functional group was prepared and it was found that the thiazole derivative (24) was the most potent and COX-2 selective compound of this class with COX-1: IC50 >100 μmol and COX-2: IC50 = 0.14 μmol on purified enzymes (Song et al., 1999).
 | |
| Fig. 7: | Conversion of indomethacin to selective COX-2 inhibitors |
 |
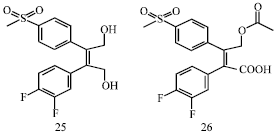
1, 2-Diarylethylene derivatives: Compounds 25 and 26 are examples of this group of compounds that is still undergoing biological testing (Black et al., 1996).
 |
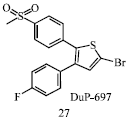
Vicinal diarylcarbocycles or heterocycles (Coxibs): These compounds represent the most important group of COX-2 inhibitors. DuP-697 (27) is the prototype of this class of compounds that is called Coxibs (Bertenshaw et al., 1995).
 |
 | |
| Fig. 8: | Conversion of nonselective COX inhibitors to COX-2 selective inhibitors |
Clinical data of 27 showed selective inhibitory activity against COX-2 (Gaus et al., 1990), but showed very long plasma half-life of 242 h in human and because of its entrahepatic recirculation, it was unacceptable for further evaluation.
All Coxibs characterized by having a central carbocyclic or heterocyclic five membered ring system bearing tow vicinal aryl moieties, such as, cyclobutenone (SC-57666) (28), pyrazole (Penning et al., 1997) (Celcoxib (celebrex®)) (29), 2 (5H) furanone (Li et al., 1999) (Refecoxib (vioxx®)) (30) and isoxazole (Talley et al., 2000a, b; Li et al., 2003) (Valdecoxib®) (31). Some Coxibs have a six-membered ring as the central heterocycle such as the pyridine derivative Etoricoxib (32) (Fig. 9).
A novel class of 6-alkylthio-substituted six membered lactone (pyranone-2-one) ring (33) has been reported to exhibit very good in vitro COX-2 inhibitory potency and selectivity (Joo et al., 2004; Kuel et al., 1984) (Fig. 8).
Structure activity relationship studies (SAR) of Coxibs showed that, substitution at position 4-of one of the aromatic ring system with a sulfonamide or a methylsulfonyl group is essential for optimum COX-2 selectivity and inhibitory potency and the presence of a p-F substituent on a non-sulfonyl vicinal phenyl ring improve in vivo activity (Venturini et al., 1998).
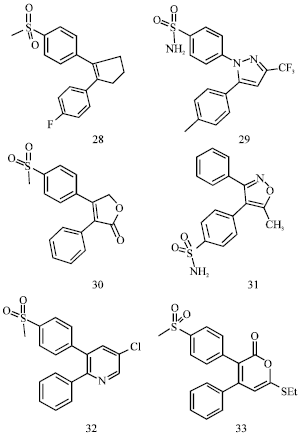 | |
| Fig. 9: | Representative examples of selective COX-2 inhibitors |
Other diarylheterocycles:
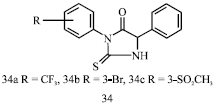
Diarylimidazolidines: Gauthier et al. (2006) reported the synthesis and molecular modelling of a new series of 3,5-diphenyl-2-thioxoimidazolidin-4-one of the general structure 34 as potential inhibitors of COX-2. It was found that derivatives 34a, 34b and 34c strongly inhibit human recombinant COX-2 at 50 μmol. Moreover COX-2 expression of these compounds was induced by LPS and the inhibitory potency was weak, this was attributed to poor aqueous stability of these thioxoimidazolidinone derivatives (Gauthier et al., 2006).
 |
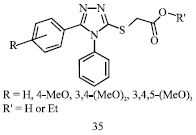
Substituted diaryl 1,2,4-triazoles: There has been a growing interest in the anti-inflammatory activity of 1,2,4-triazole derivatives, of these compounds; the 4,5-diaryl-1,2,4-triazolthioacetic acid and their ethyl esters 35 which displayed good in vivo anti-inflammatory activity against carrageenin-induced rat paw oedema (Maxwell et al., 1984).
 |
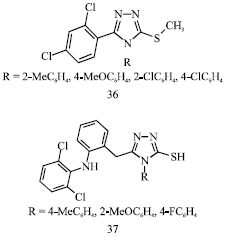
In addition to the methylthioether derivatives 36 and the 2,6-dichloroanilinobenzyl derivatives 37 which produced good anti-inflammatory activity (Gosowami et al., 1984; Amir and Shikla, 2004).
 |
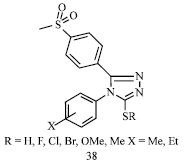
Recently Navidapour et al. (2006) designed and synthesized a new type of 4,5-diaryl-4H-1,2,4-triazole, possessing C-3 thio and alkylthio substituents of the structure 38 for evaluation as selective COX-2 inhibitors. It was reported that compound 3-ethylthio-5-(4-flurophenyl)-4-(4-methylsulfonylphenyl)-4H-1,2,4-triazole exhibited a high in vitro selectivity and showed good antiinflammatory activity compared to celcoxib in a carrageenan-induced rat paw edema assay (Navidapour et al., 2006).
 |
MOLECULAR MODELLING STUDIES
The molecular modeling studies linked between the pharmacological effect and chemical structure which shed more light on structure activity relationship (Al-Rashood et al., 2006; Abou-Zeid, 2002; Abou-Zeid and El-Mowafy, 2002; Abou-Zeid et al., 2007; Goda et al., 2008).
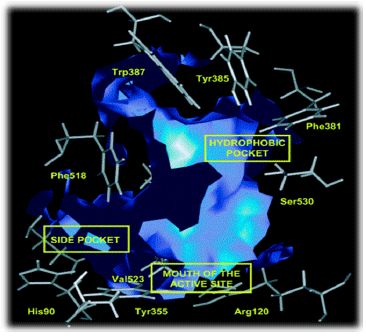 | |
| Fig. 10: | Different region of COX-1 active site (Al-Turki, 2010) |
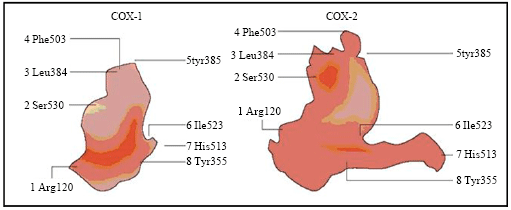 | |
| Fig. 11: | The left hand shows a schematic diagram of the binding site of COX-1. The right hand panel depicts the COX-2 binding site Residues(1-8) represents the key amino acid of both COX enzymes (Al-Turki, 2010) |
Inhibition of COX enzyme at the molecular level is mediated through the blockade of AA to access the COX active site. Accordingly, comparative molecular modelling studies of several selective and non selective NSAIDs in complex with COX-1 and COX-2 have been done to delineate features that differentiate their mode of interaction with COX-1 and COX-2 (Juni et al., 2002). The molecular modelling will be discussed in section 3.
Structure of COX binding sites: The reported molecular modelling studies based on the x-ray crystallography of the 3-D structures of COX-1 and COX-2 indicated that COX-1 and COX-2 are 63% identical and 77% similar at the amino acid level (Al-Turki, 2010; Fabiola et al., 2001) (Fig. 10).
COX binding site can be considered as a hydrophobic channel extending from the membrane binding domain. In the upper part of the channel, both isozyme possess a Ser 530 which is the amino acid acetylated by aspirin, whereas Tyr 385 located at top of the channel.
The main difference between the two COX active sites is the replacement of the relatively bulky isoleucin (Ile) residue in COX-1 by Valine (Val) at position 523 of the active site of the enzyme. This substitution opens a polar side pocket, enlarging the volume of COX-2 active site and giving access to Arg 513 replaced in COX-1 by a histidine at the same position (Fig. 11). This will cause a structural modification in COX-2 enzyme that allows access to an additional side pocket which is a prerequisite for COX-2 drug selectivity.
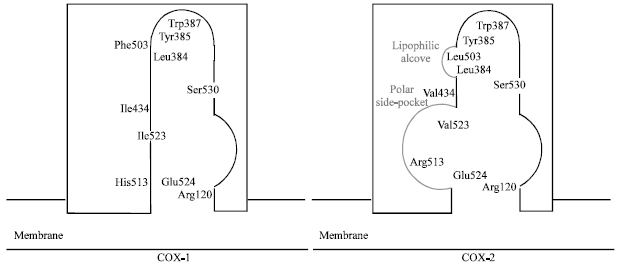 | |
| Fig. 12: | Schematic representation of the active site of the two isoenzymes COX-1 and COX-2 (Al-Turki, 2010) |
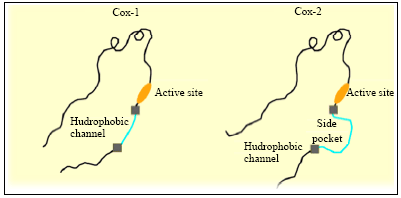 | |
| Fig. 13: | Structure difference between COX-1 and COX-2 enzymes |
Second, in the apex of the COX-2 binding site, substitution of Phe 503 in COX-1 by Leu 503 generates a small aclove which is hydrophobic due to the presence of Leu 384, Tyr 385 and Tryp 387 (Fig. 12).
Thus, there is still a need for novel, selective and potent COX-2 inhibitors with an improved profile compared to current COX-2 inhibitors.
CONCLUSIONS
The discovery of the existence of two cyclooxygenases greatly refines the understanding of how the therapeutic and toxic effect of NSAIDs relate to inhibition of PG synthesis.
As was reported, NSAIDs inhibit both COX-1 and COX-2 to different extents. This accounts for their anti-inflammatory and analgesic activities and also their unwanted GI side effect (Carabaza et al., 1996).
Current evidence indicates that selective COX-2 inhibitors have important adverse cardiovascular effects that include increased risk for myocardial infarction, stroke, heart failure and hypertension (Antman et al., 2007).
Thus, the development of selective COX-2 inhibitors could be a big step ahead in the therapeutic treatment of anti-inflammatory diseases with fewer risks and side effects (Al-Turki, 2010). As shown in this study, the selective COX-2 diaryl heterocyclic, Coxib group; is characterized by having different 1,2-diaryl five-membered or six-membered heterocycles that are considered as pharmacophore templates such as, Celcoxib (29), Refecoxib (30), Valdecoxib (31) and Etoricoxib (32) (Riendeau et al., 2002; Friesen et al., 1996; Prasit et al., 1999).
Moreover, the SAR studies have shown that the substituted sulfonyl group present in the structure of Coxibs, is considered one of the pharmacophoric moieties responsible for the selective recognition with the key amino acid residues at COX-2 active site pocket.
Also, it has been reported that compounds having aryl methylsufone or aryl sulfonamide moieties display a propensity for COX-2 selectivity (Smith et al., 2000).
Furthermore, taking in consideration the main difference between the two COX active sites which is the replacement of Ile523 in COX-1 by the less bulky Val523 in COX-2 active site, results in opening a polar side pocket that enlarges the volume of COX-2 active site and is considered a prerequisite for COX-2 drug selectivity (Mengle-Gaw and Schwartz, 2002) (Fig. 13).
REFERENCES
- Abou-Zeid, L.A., A.M. El-Mowafy, D. Eikel, H. Nau and M. El-Mazar, 2007. Computational characteristics of valproic acid binding to Histone deacetylase. Saudi Pharm. J., 15: 183-189.
Direct Link - Abou-Zeid, L.A. and A.M. El-Mowafy, 2002. Molecular dynamics simulation characteristics of resveratrol interaction with human estrogen receptor-α: Distinct recognition from diethylstilbestrol. J. Mol. Struct. Theochem., 593: 39-48.
CrossRef - Al-Rashood, S.T., I.A. Aboldahab, M.N. Nagi, L.A. Abou-zeid and A.M. Abdel-Aziz et al., 2006. Synthesis, dihydrofolate reductase inhibition, antitumor testing and molecular modeling study of some new 4(3H)-quinazolinone analogs. Bioorg. Med. Chem., 15: 8608-8621.
PubMed - Amir, M. and K. Shikha, 2004. Synthesis and anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities of some new 2-[(2,6-dichloroanilino) phenyl] acetic acid derivatives. Eur. J. Med. Chem., 39: 535-545.
CrossRefPubMedDirect Link - Antman, E.M., J.S. Bennett, A. Daugherty, C. Furberg, H. Roberts and K.A. Taubert, 2007. Use of nonsteroidal antiinflammatory drugs. An update for clinicians: A scientific statement from the american heart association. Circulation, 115: 1634-1642.
Direct Link - Dannhardt, G. and W. Kiefer, 2001. Cyclooxygenase inhibitors current status and future prospects. Eur. J. Med. Chem., 36: 109-126.
PubMed - Bertenshaw, S.R., J.J. Talley, D.J. Rogier, M.J. Graneto and R.S. Rogers et al., 1995. E. 3,4-Diarylthiophenes are selective COX-2 inhibitors. Bioorg. Med. Chem. Lett., 5: 2919-2922.
CrossRef - Black, W.C., C. Bayly, M. Belley, C.C. Chan and S. Charleson et al., 1996. From indomethacin to a selective COX-2 inhibitor: Development of indolalkanoic acids as potent and selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett., 6: 725-730.
CrossRefDirect Link - Carabaza, A., F. Cabre, E. Rotllan, M. Gomez, M. Gutierrez, L. Garcıa and D. Mauleon, 1996. Stereoselective inhibition of inducible cyclooxygenase by chiral nonsteroidal anti-inflammatory drugs. J. Clin. Pharmacol., 36: 505-512.
Direct Link - Saari, W.S. and S.W. King, 1973. Synthesis and biological activity of (6aS)-10,11-dihydroxyaporphine, the optical antipode of apomorphine. J. Med. Chem., 16: 171-172.
PubMed - Crofford, L.J., P.E. Lipsky, P. Brooks, S.B. Abramson, L.S. Simon and L.B.A. Putte, 2000. Basic biology and clinical application of cyclooxygenase-2 inhibitors. Arthritis Rheum., 43: 4-13.
3.0.CO;2-V/pdf target='_blank' class='btn btn-sm btn-outline-primary mr-3 mt-3'>Direct Link - Chrischilles, E.A. and R.B. Wallace, 1993. Nonsteroidal anti-inflammatory drugs and blood pressure in an elderly population. J. Gerontol., 48: M91-M96.
Direct Link - Dannenberg, A.J., N.K. Altorki, J.O. Boyle, C. Dang, L.R. Howe, B.B. Weksler and K. Subbaramaiah, 2001. Cyclooxygenase 2: A pharmacological target for the prevention of cancer. Lancet Oncol., 2: 544-551.
PubMed - Dannhardt, G. and S. Laufer, 2000. Structural approaches to explain the selectivity COX-2 inhibitors: Is there a common pharmacophore. Curr. Med. Chem., 7: 1101-1112.
PubMed - Davis, R. and R.N. Brogden, 1994. Nimesulide. An update of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy. Drugs, 48: 431-454.
PubMed - Dewitt, D.L. and W.L. Smith, 1988. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc. Natl. Acad. Sci., 85: 1412-1416.
PubMedDirect Link - Dubois, R.N., S.B. Abramson, L. Crofford, R.A. Gupta, L.S. Simon, L.B.V.D. Putte and P.E. Lipsky, 1998. Cyclooxygenase in biology and disease. FASEB J., 12: 1063-1073.
CrossRefPubMedDirect Link - Friesen, R.W., D. Dube, R. Fortin, R. Frenette and S. Prescott et al., 1996. Novel 1,2-diarylcyclobutenes: Selective and orally active COX-2 inhibitors. Bioorg. Med. Chem. Lett., 6: 2677-2682.
CrossRef - Gaus, K.R., W. Galbraith, R.J. Roman, S.B. Haber and J.S. Kerr et al., 1990. Anti-inflammatory and safety profile of Dup 697, a novel orally effective prostaglandin synthesis inhibitor. J. Pharmacol. Exp. Ther., 254: 180-187.
PubMed - Gauthier, M.P., C. Michaux, S. Rolin, C. Vastersaegher and X. de Leval et al., 2006. Synthesis, molecular modelling and enzymatic evaluation of (±) 3,5-diphenyl-2-thioxoimidazolidin-4-ones as new potential cyclooxygenase inhibitors. Bioorg. Med. Chem., 14: 918-922.
PubMed - Goda, F.E., A.S. Tantawy, S., L.A. Abou-zeid, S.M. Badr and K.B. Selim, 2008. Synthesis, biological evaluation and molecular modeling investigation of some new benzimidazole analogs as antiviral agents. Saudi Pharm. J., 16: 103-111.
Direct Link - Gosowami, B.N., J.C.S. Kataky and J.N. Baruah, 1984. Synthesis and antibacterial activity of 1-(2,4-dichlorophenyl)-4-substituted thiosemicarbazides, 1,2,4-triazoles and their methyl derivatives. J. Heterocycl. Chem., 21: 1225-1229.
Direct Link - Graumlich, J.F., 2001. Preventing gastrointestinal complications of NSAIDs: Risk factors, recent advances and latest strategies. Postgrad Med., 109: 117-128.
PubMedDirect Link - Griffin, M.R., J.M. Piper and J.R. Daugherty, M. Snowden and W.A. Ray, 1991. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann. Intern. Med., 114: 257-263.
PubMed - Handel, J. and O.H. Nielsen, 1997. Expression of cyclooxygenase-2 mRNA in active inflammatory bowel disease. Am. J. Gastroenterol., 92: 1170-1173.
Direct Link - Hollander, D., 1994. Gastrointestinal complications of nonsteroidal anti-inflammatory drugs: Prophylactic and therapeutic strategies. Am. J. Med., 96: 274-281.
Direct Link - Houston, A.M. and S.J. Tech, 2004. COX-2 inhibitors: A review. Ped. Emerg. Care, 20: 396-399.
Direct Link - Hoozemans, J.J., R. Veerhuis, A.J. Rozemuller and P. Eikelenboom, 2003. Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer's disease. Curr. Drug Targets, 4: 461-468.
PubMed - Huff, R., P. Collins, S. Kramer, K. Seibert, C. Koboldt, S. Gregory and P. Isakson, 1995. A structural feature of N-[2-(cyclohexyloxy)-4-nitrophenyl] methanesulfonamide (NS-398) that governs its selectivity and affinity for cyclooxygenase 2 (COX2). Inflamm. Res., 44: 145-146.
Direct Link - Johnson, A.G., L.A. Simons, J. Simons, Y. Friedlander and J. McCallum, 1993. Non-steroidal anti-inflammatory drugs and hypertension in the elderly: A communitybased cross-sectional study. Br. J. Clin. Pharmacol., 35: 455-459.
Direct Link - Joo, Y.H., J.K. Kim, S.H. Kang, M.S. Noh and J.Y. Ha et al., 2004. 2,3-Diarylpyran-4- ones: A new series of selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett., 14: 2195-2202.
CrossRef - Juni, P., A.W. Rutjes and P.A. Dieppe, 2002. Are selective COX 2 inhibitors superior to traditional non steroidal anti-inflammatory drugs. Br. Med. J., 324: 1287-1288.
Direct Link - Fabiola, G.F., L. Damodharan, V. Pattabhi and K. Nagarajan, 2001. Cyclooxygenase-2, An attractive target for fruitful drug design. Curr. Sci., 80: 26-34.
Direct Link - Kalgutkar, A.S., A.B. Marnett, B.C. Crews, R.P. Remmel and L.J. Marnett, 2000. Ester and amide derivatives of the Nonsteroidal Antiinflammatory drug, Indomethacin, as selective COX-2 inhibitors. J. Med. Chem., 43: 2860-2870.
Direct Link - Kalgutkar, A.S., B.C. Crews, S.W. Rowlinson, A.B. Marnett, K.R. Kozak, R.P. Remmel and L.J. Marnett, 2000. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: Facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci., 97: 925-937.
Direct Link - Kalgutkar, A.S., B.C. Crews, S.W. Rowlinson, C. Garner, K. Seibert and L.J. Marnett, 1998. Aspirin-like molecules that covalently inactivate cyclooxygenase-2. Science, 280: 1268-1270.
Direct Link - Leahy, K.M., A.T. Koki and J.L. Masferrer, 2000. Role of cyclooxygenases in angiogenesis. Curr. Med. Chem., 7: 1163-1170.
Direct Link - Kawaguchi, H., C.C. Pilbeam, G. Gronowicz, C. Abreu and B.S. Fletcher et al., 1995. Transcriptional induction of prostaglandin G/H synthase-2 by basic fibroblast growth factor. J. Clin. Invest., 96: 923-930.
Direct Link - Kuel, F.A., H.W. Daugherty and E.A. Ham, 1984. Interactions between prostaglandins and leukotrienes. Biochem. Pharmacol., 33: 1-5.
PubMed - Laine, L., 1996. Nonsteroidal anti-inflammatory drug gastropathy. Gastrointest. Endosc. Clin. N. Am., 6: 489-504.
PubMed - Simon, L.S., 2001. COX-2 inhibitors. Are they nonsteroidal anti-inflammatory drugs with a better safety profile. Gastroenterol. Clin. N. Am., 30: 1011-1025.
PubMed - Therien, M., C. Brideau, C.C. Chan, W.A. Cromlish and J.Y. Gauthier et al., 1996. Synthesis and biological evaluation of 5,6-diarylimidazo(2,1-b) thiazole as selective COX-2 inhibitors. Bioorg. Med. Chem. Lett., 7: 47-52.
CrossRef - Li, C.S., C. Soucy-Breau and N. Ouimet, 1995. Improved Synthesis of a Selective COX-2 Inhibitor, 6-(2,4-Difluorophenoxy)-5-methanesulfonamidoindan-1-one (Flosulide). Synthesis, 10: 1355-1356.
CrossRefDirect Link - Li, C.S., W.C. Black, C. Brideau, C.C. Chan and S. Charleson et al., 1999. A new structural variation on the methanesulfonylphenyl class of selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett., 9: 3181-3186.
PubMed - Li, C.S., C. Brideau, C.C. Chan, C. Savoie and D. Claveau et al., 2003. Pyridazinones as selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett., 13: 597-600.
CrossRef - Masferrer, J.L., K.M. Leahy, A.T. Koki, B.S. Zweifel and S.L. Settle et al., 2000. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res., 60: 1306-1311.
PubMedDirect Link - MacDonald, T.M., S.V. Morant, G.C. Robinson, M.J. Shield, M.M. McGilchrist, F.E. Murray and D.G. McDevitt, 1997. Association of upper gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs with continued exposure: Cohort study. Br. Med. J., 315: 1333-1337.
Direct Link - Maxwell, J.R., D.A. Wasdahl, A.C. Wolfson and V.I. Stenberg, 1984. Synthesis of 5-aryl- 2H-tetrazoles, 5-aryl-2H-tetrazole-2-acetic acids and [(4-phenyl-5-aryl-4H-1,2,4-triazol-3-yl)thio]acetic acids as possible superoxide scavengers and anti-inflammatory agents. J. Med. Chem., 27: 1565-1570.
PubMed - Mengle-Gaw, L.J. and B.D. Schwartz, 2002. Cyclooxygenase-2 inhibitors: Promise or peril. Mediators Inflamm., 11: 275-286.
PubMed - Meyerson, B.A. and B. Linderoth, 2006. Mode of action of spinal cord stimulation in neuropathic pain. J. Pain Symptom Manage., 31: S6-S12.
Direct Link - Navidapour, L., H. Shafaroodi, K. Abdi, M. Amini, M.H. Ghahremani, A.R. Dehpour and A. Shafiee, 2006. Design, synthesis and biological evaluation of substituted 3-alkylthio-4,5-diaryl-4H-1,2,4-triazoles as selective COX-2 inhibitors. Bioorg. Med. Chem., 14: 2507-2517.
CrossRefPubMedDirect Link - Noble, S.L., D.S. King and J.I. Olutade, 2000. Cyclooxygenase-2 enzyme inhibitors: Place in therapy. Am. Fam. Physician, 61: 3669-3676.
PubMed - Patrignani, P., M.R. Panara, A. Greco, O. Fusco and C. Natoli et al., 1994. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthase. J. Pharmacol. Exp. Ther., 271: 1705-1712.
Direct Link - Penning, T.D., J.J. Talley, S.R. Bertenshaw, J.S. Carter and P.W. Collins et al., 1997. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase 2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J. Med. Chem., 40: 1347-1365.
PubMed - Pope, J.E., J.J. Anderson and D.T. Felson, 1993. A meta-analysis of the effects of nonsteroidal anti-inflammatory drugs on blood pressure. Arch. Intern. Med., 153: 477-484.
PubMed - Whelton, A., 1999. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: Physiologic foundations and clinical implications. Am. J. Med., 106: 13S-24S.
Direct Link - Prasit, P., Z. Wang, C. Brideau, C.C. Chan and S. Charlson et al., 1999. The discovery of rofecoxib, [MK 966, Vioxx, 4-(4'-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2-inhibitor. Bioorg. Med. Chem. Lett., 9: 1773-1778.
CrossRef - Rodriguez, L., C. Cattaruzzi, M.G. Troncon and L. Agostinis, 1998. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists and other antihypertensive drugs. Arch. Intern. Med., 39: 35-46.
Direct Link - Scheiman, J.M., 1996. NSAIDs, gastrointestinal injury and cytoprotection. Gastroenterol. Clin. North Am., 25: 279-298.
PubMed - Shiff, S.J., P. Shivaprasad and D.L. Santini, 2003. Cyclooxygenase inhibitors: Drugs for cancer prevention. Curr. Opin. Pharmacol., 3: 352-361.
CrossRef - Smith, W.L., 1992. Prostanoid biosynthesis and mechanisms of action. Am. J. Physiol. Renal. Physiol., 263: F181-F191.
Direct Link - Vane, J.R., 1971. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol., 231: 232-235.
PubMedDirect Link - Smith, W.L., D.L. DeWitt and R.M. Garavito, 2000. Cyclooxygenases: Structural, cellular and molecular biology. Ann. Rev. Biochem., 69: 145-182.
PubMed - Song, Y., D.T. Connor, R. Doubleday, R.J. Sorensen and A.D. Sercel et al., 1999. Synthesis, structure−activity relationships and in vivo evaluations of substituted di-tert-butylphenols as a novel class of potent, selective and orally active cyclooxygenase-2 inhibitors. 1. thiazolone and oxazolone series. J. Med. Chem., 42: 1151-1160.
CrossRef - Sperling, R.I., 1995. Eicosanoids in rheumatoid arthritis. Rheum. Dis. Clin. North Am., 21: 741-758.
PubMed - Talley, J.J., D.L. Brown, J.S. Carter, M.J. Graneto and C.M. Koboldt et al., 2000. 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, valdecoxib: A potent and selective inhibitor of COX-2. J. Med. Chem., 43: 775-777.
CrossRefDirect Link - Teismann, P., K. Tieu, D.K. Choi, D.C. Wu and A. Naini et al., 2003. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Natl. Acad. Sci. USA., 100: 5473-5478.
PubMed - Flower, R.J. and J.R. Vane, 1972. Inhibition of prostaglandin synthetase in brain explains the antipyretic activity of paracetamol (4-acetamidophenol). Nature, 240: 410-411.
Direct Link - Venturini, C.M., P. Isakson and P. Needleman, 1998. Non steroidal anti-inflammatory drug-induced renal failure. Curr. Opin. Nephrol. Hypertens., 7: 79-82.
PubMed - Wu, G., R.J. Kulmacz and A.L. Tsai, 2003. Cyclooxygenase inactivation kinetics during reaction of prostaglandin H synthase-1 with peroxide. Biochemistry, 42: 13772-13777.
PubMed