
Anurekha Jain
B.R. Nahata College of Pharmacy, Mandsaur (MP), India
S.C. Chaturvedi
School of Pharmacy, Devi Ahilya Vishav Vidalaya, Indore, India
Asian Journal of Biochemistry
Year: 2008 | Volume: 3 | Issue: 5 | Page No.: 330-336







ABSTRACT
Various lead structures of the compounds of angiotensin II antagonist are reported in the literature. Studying the Structure-Activity Relationships (SAR) for such compounds has been a fascination for scientists and efforts have been made to identify the essential physico-chemical requirements for the angiotensin type 1 (AT1) receptor selective, angiotensin type 2 (AT2) receptor selective and some AT1 and AT2 balanced antagonistic activity compounds. With an aim to identify the structural requirements for selective AT1 activity, a Quantitative SAR (QSAR) analysis was carried out on a series of benzimidazole derivative bearing acidic heterocycle AII receptor antagonists. The QSAR expressions were generated using 19 compounds and the predictive ability of the resulting model was evaluated against a test set of 7 compounds. The internal (cross validated squared correlation coefficient) and external consistency (predictive correlation coefficient) of the QSAR model was 0.895 and 0.405, respectively. Analysis of result from the present QSAR study indicates that geometrical, structural and shape descriptors govern the angiotensin II AT1 inhibitory activity.
PDF Abstract XML References Citation
How to cite this article
Anurekha Jain and S.C. Chaturvedi, 2008. Rationalization of Physicochemical Property of Some Substituted Benzimidazole Bearing Acidic Heterocyclic Towards Angiotensin II Antagonist: A QSAR Approach. Asian Journal of Biochemistry, 3: 330-336.
DOI: 10.3923/ajb.2008.330.336
URL: https://scialert.net/abstract/?doi=ajb.2008.330.336
DOI: 10.3923/ajb.2008.330.336
URL: https://scialert.net/abstract/?doi=ajb.2008.330.336
INTRODUCTION
The Renin Angiotensin Aldosterone System (RAAS) is a proteolytic cascade that plays an important role in electrolyte homeostasis and in the regulation of blood pressure, but it is also involved in the pathogenesis of hypertension and renal disease. The Angiotensin Converting Enzyme (ACE) to produce the octapeptide angiotensin II (AII) which the main effectors hormone of the RAAS.
The RAAS begins with the release of the aspartic protease renin from the juxtaglomerular cell of the kidney. This enzyme is responsible for the conversion of angiotensinogen to the inactive decapeptide angiotensin I. In turn, angiotensin I is cleaved by the angiotensin-converting enzyme to produce the octapeptide angiotensin II (AII), which is the main effector hormone of the RAAS.
AII is the major regulator of the blood pressure, electrolyte balance and endocrine function related to cardiovascular disease such as hypertension. More ever, it has been shown that AII plays a role in the various pathological situations involving tissue remodeling, such as cardiac hypertrophy. Recent finding indicate the involvement of this peptide also in cancer (Juillerat-Jeanneret et al., 2004; Fogarty et al., 2002).
AII affect most of the biological function by activating selective membrane–bound receptor. Two distinct subtypes of AII receptor [type 1(AT1) and type 2(AT2)] have been identified and both belongs to the G protein-coupled receptor superfamily. (GPCRs). AT1 and AT2 are seven- transmembrane-spanning receptors, comprising an extracellular glycosylated region connected to the seven transmembrane alfa-helices, which are linked by three intracellular and three extracellular loops. The carboxy-terminal domain of the protein is cytoplasmic and is a regulatory site. AT1 is a 359-amino acid protein, while AT2 is made up of 363 amino acid and is 30% homologous with AT1; both receptors are N-linked glycosylated post-translationally.
AT1 receptor are expressed in various parts of the body and mediate all of the known effects associated with AII, such as vasoconstriction, aldosterone release and other function that tend to elevate blood pressure and cause hypertrophy and hyperplasia of target cells. The role AT2 receptor is less fully understood.
Tiziano et al. (2006) recently studied 3D-QSAR model was calculated based on the alignment obtained by docking several ligands into the AT1 receptor. Given the important role played by the RAAS in hypertension, this system is the main target of any effective therapy, the first choice class of drugs to influence the RAAS targeting is that of ACE inhibitors.
These drugs block the formation of A II and also prevent the conversion of bradykinin to inactive peptides. Although bradykinin may contribute to the beneficial effects of Ace inhibitor through its vesorelaxing effects, its accumulation determines some disadvantages such as the development of coughing and angiodema, which are side effect often associated with ACE-inhibitor therapy. Moreover, ACE inhibitor do not completely suppress AII, because its formation ensured also by the ACE-independent pathways. For these reasons, it was particularly important when A II subtype 1 receptor antagonists were developed as a new class of antihypertensive agents clinically because of lesser side effects and better therapeutics profile than ACE inhibitors.
The QSAR analysis of Angiotensin II Antagonist is the current highly interested area of research in this area (Pandya et al., 2001; Pandya and Chaturvedi, 2005).
To gain insight into the structural and molecular requirement influencing the angiotensin II antagonistic activity, we herein describe QSAR analysis of Benzimidazole derivative bearing acidic heterocycles. The relevance of the model for the design of novel derivatives should be assessed not only in terms of predictivity, either internal or external, but also in terms of their ability to provide a chemical and structural explanation of their binding interaction. Here, we propose a general model for the antagonist and present minimal structural requirement for an Angiotensin II antagonist. These results should serve as a guideline in design of more potent and selective AII antagonist.
MATERIALS AND METHODS
The A-II receptor antagonistic activity data of Benzimidazole derivative bearing acidic heterocycles were taken from the reported study Kohara et al. (1996) (Table 1). The biological activity data (IC50 in 10-7 M) was converted to negative logarithmic mole dose (pIC50) for Quantitative Structure Activity Relationship (QSAR) analysis. The molecular study was performed using CS ChemOffice (CS Chem Office version 8 Cambridge soft version) and dragon (Todeschini and Consonni, 2001) program while the regression analysis was carried on the VALSTAT (Gupta et al., 2004).
The molecular structures of twenty-Six compounds (except compounds with % inhibitory activity) were sketched using Chem Draw and transferred to ChemUltra to convert them into 3D structures. The energy minimization of the molecule was done using molecular mechanics (MM2) until the RMS gradient value became smaller than 0.1 kcal mol-1 Å. The energy minimized molecules were subjected to the re-optimization via Austin model-1 (AM1) method until the RMS gradient attained a value smaller than 0.0001 kcal mol-1 Å using MOPAC. The geometry optimization of the lowest energy structure was carried out using EF routine. The descriptor values for the entire molecule were calculated using compute properties module of the program. The molecule was saved as MOL file format. Pursuly, the MOL file was used for calculation of various physicochemical properties using Dragon program. The data was transferred to the statistical program in order to establish a correlation between physicochemical parameters as independent variables and pIC50 as dependent variable employing sequential multiple linear regression analysis method. In sequential multiple linear regression, the program searches all the permutation and combination sequentially for the data set. The Values of descriptors, which are significant in equation, are shown in Table 1. Series was divided into training set of 19 compounds and test set of 7 compounds on the basis of structural diversity and cover the complete range of variation in antagonist activity. The ±data within the parentheses are the standard deviation, associated with coefficient of descriptors in regression equations. The best model was selected from the various statistically significant equations on the basis of observed squared correlation coefficient (r2), Standard Error of Estimate (SEE), sequential Fischer test (F), bootstrapping squared correlation coefficient (r2bs), bootstrapping standard deviation (Sbs), cross validated squared correlation coefficient using leave one out procedure (Q2), chance statistics (evaluated as the ratio of the equivalent regression equations to the total number of randomized sets; a chance value of 0.001 corresponds to 0.1% chance of fortuitous correlation), outliers (on the basis of Z-score value) and predictive squared correlation coefficient of test set (r2pred).
| Table 1: | Structure, activities and physicochemical properties of Benzimidazole derivative bearing acidic heterocycles used in training and test set |
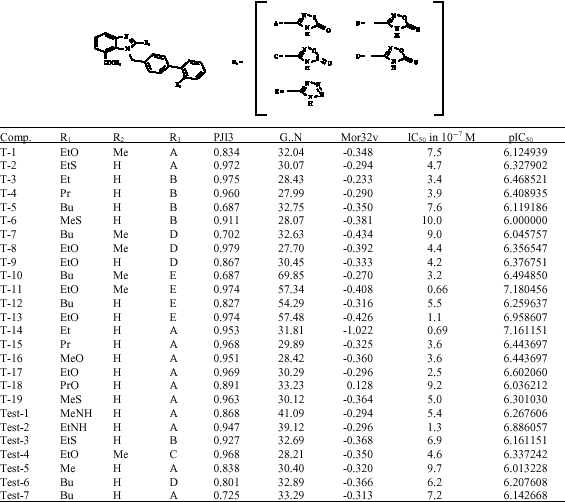 | |
RESULTS AND DISCUSSION
Training set of 19 compounds was subjected to stepwise multiple linear regression analysis, in order to develop QSAR between antagonistic activity at AII AT1 receptor as dependent variables and substituents constants as independent variables, several significant equations were obtained (Eq. 1-5).
pIC50 = 3.047 (±0.448) ASP+0.013 (±0.003) G (N..N)+0.625 (±0.108) Mor15m +4.044 n = 19, r = 0.924, r2 = 0.853, SEE = 0.145, F = 29.019 | (1) |
pIC50 = 4.177 (±0.581) ASP+0.020 (±0.007) Mor02u -0.388 (±0.079) Mor09m+3.391 n = 19, r = 0.910, r2 = 0.828, SEE = 0.157, F = 24.107 | (2) |
pIC50 = 3.268 (±0.508) ASP +0.0142 (±0.003) G (N..N)+1.032 (±0.209) Mor15v +3.642 n = 19, r = 0.904, r2 = 0.818, SEE = 0.162, F = 22.488 | (3) |
pIC50= 0.232 (±0.043) Mor05u -0.343 (±0.085) Mor09m+1.404 (±0.261) Mor26m +8.252 n = 19, r = 0.902, r2 = 0.813, SEE = 0.164, F = 21.778 | (4) |
pIC50 = 1.984 (±0.408) PJI3+0.016 (±0.003) G (N..N) -0.886 (±0.198) Mor32v+3.738 n = 19, r = 0.896, r2 = 0.803, SEE = 0.168, F = 20.333 | (5) |
Training set was used to explore conformational and geometrical related physiochemical properties that are helpful in understanding the probable binding site of drug with enzyme. Correlation were established between physicochemical parameters and Angiotensin antagonistic activity using sequential multiple linear regression technique. Several significant equations with coefficient of correlation (r~0.900) were obtained.
Only high correlation coefficient is not enough to select the equation as a model and hence various statistical approaches were used to confirm the robustness and practical applicability of equations. The Eq. 1-5 tested for presence of outlier. Equation 1-5 showed probability of chance correlation is less than 0.1% in randomize biological activity test. Bootstrapping technique were employed to confirmed the contribution of physiochemical properties of the molecules to the activity were equi-intense or of different rank. The value of bootstrapping squared correlation coefficient and bootstrapping standard deviation implies that the equations are proper representative of the group of analogs (Table 2).
The internal consistency of the training set was confirmed using leave one out (loo) cross validation method to ensure the robustness of the equations. Equations showed good internal consistency (Q2 = 0.688-0.768), which reduces the probability of coincidental correlation of the expression. Expression which having significant internal consistency may not be applicable for the analogs, which were never used in generation of correlation and therefore, the predictive power of Eq. 1-5 were further confirmed by test set of five compounds. Equation 1-4 showed poor predictivity of test set with low r2pred value (<0.243). Therefore equation 5 was considered as model.
The model has better correlation coefficient (r = 0.895), which accounts for more than 80.0% of the variance in the activity, also the inter-correlation among the parameters is less than 0.316 (Table 3). The equation shows, that in multi-variant model, dependent variable can be predicted from a linear combination of the independent variables. The p-value is less than 0.01 for each physiochemical parameters involved in model generation. The data showed overall internal statistical significance level better than 99.9% as it exceeded the tabulated F(3,15 α 0.001) = 10.8. The model was further tested for outlier by Z-score method and no compound was found to be an outlier (Table 2) which suggested that the model is able to explain the structurally diverse analogs that is helpful in designing of more potent compounds using physiochemical parameters. Leave one out cross validation method was employed for prediction of the activity (Fig. 1), cross-validated squared correlation co-efficient (Q2 = 0.672), predictive residual sum of square (SPRESS = 0.217) and standard error of prediction (SDEP = 0.192) suggested good internal consistency as well as predictive ability of the biological activity with low SDEP. The r2bs is at par with the conventional squared correlation coefficient (r2). Randomized biological activity test (Chance <0.001) revealed that the results were not based on chance correlation. The robustness and wide applicability of the model was further explained by significant r2pred value (0.405) of test set data (Fig. 2). In general the model fulfills the statistical validation criteria in a significant echelon to achieve theoretical base for proposing more active compounds. In the model biological activity contributed positively by PJI3 and G (N..N) While negatively contributed by Mor32v.
| Table 2: | QSAR statistics of significant equations |
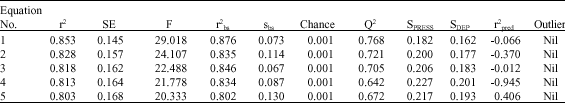 | |
| Table 3: | Inter-correlation matrix of descriptors used in model |
 | |
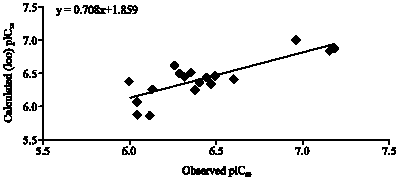 | |
| Fig. 1: | A graphical representation of observed pIC50 and calculated (loo) pIC50 from Eq. 5 |
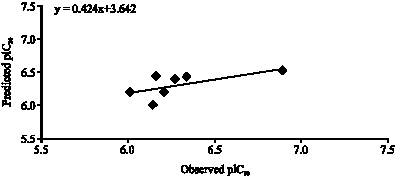 | |
| Fig. 2: | A graphical representation of observed pIC50 and predicted pIC50 from Eq. 5 |
PJI3 (Bath et al., 1995) is three dimensional petijean shape index and based on the geometry of the molecule in space. The PJI3 contribution suggested that the specific orientation of the molecule is decisive for interaction with receptor.
G (N..N) (Balaban, 1997) is conformational dependent descriptor and based on the geometry of the molecule and G (N..N) is sum of geometrical distance between nitrogen and nitrogen in the molecule and help in proposing possible pharmacophoric character for a drug receptor interaction. The G (N..N) Contributions revealed that the nitrogen-nitrogen distance in space is crucial for the hydrogen bond interaction with receptor and might be govern proper orientation to the molecules.
Mor32v (Gasteiger et al., 1996; Schuur et al., 1996; Schuur and Gasteiger, 1997; Todeschini and Consonni , 2000) is a Morse code-signal 32 weighted by atomic Vander Waals volumes. The Morse code (3D molecular representation of structure based on electron diffraction code) was calculated by summing atom weights viewed by a different angular scattering function. The values of these code functions were calculated at 32 evenly distributed values of scattering angle(s) in the range of 0-31 Å-1 from the three dimensional atomic co-ordinates of a molecule. The 3D-MoRSE code calculated using following expression:
| Where: |
| s | = | Scattering angle |
| rij | = | Interatomic distance of ith and jth atom |
| Ai and Aj | = | Atomic properties of ith and jth atom respectively including atomic number, atomic mass, partial atomic charge, residual electro- negativities and atom polarizability. |
The contribution of MoRSE code suggested that the Vander Waals volume is decisive in the interaction with receptor.
The results and discussion made above indicate that geometric descriptors and MoRSE code of AT1 receptor angiotensin II antagonist activity of benzimidazole derivative bearing acidic heterocycles can be modeled excellently.
ACKNOWLEDGMENTS
We authors are thankful to the HOD, School of Pharmacy, DAVV, Indore for providing facilities and support to complete this research.
REFERENCES
- Balaban, A.T., 1997. From chemical topology to 3D geometry. J. Chem. Inform. Comput. Sci., 37: 645-650.
CrossRefDirect Link - Bath, P.A., A.R. Poirrette, P. Willett and F.H. Allen, 1995. The extent of the relationship between the graph-theoretical and the geometrical shape coefficients of chemical compounds. J. Chem. Inform. Comput. Sci., 35: 714-716.
CrossRef - Gasteiger, J., J. Sadowski, J. Schuur, P. Selzer, L. Steinhauer and V. Steinhauer, 1996. Chemical information in 3D space. J. Chem. Inform. Comput. Sci., 36: 1030-1037.
CrossRef - Gupta, A.K., B.M. Arokia and S.G. Khakhedikar, 2004. VALSTAT: Validation program for quantitative structure activity relationship studies. Indian J. Pharm. Sci., 66: 396-402.
Direct Link - Pandya, T., S.K. Pandey, M. Tiwari, S.C. Chaturvedi and A.K. Saxena, 2001. 3-D QSAR studies of triazolinone based balanced AT1/AT2 receptor antagonists. Bioorg. Med. Chem., 9: 291-300.
CrossRefPubMedDirect Link - Pandya, T. and S.C. Chaturvedi, 2005. Structure-activity relationship study of some triazolinone based compounds with antagonistic balanced activity on angiotensin II receptor subtypes AT1 and AT2. A three-dimensional quantitative structure-activity relationship investigation. Arzneimittelforschung, 55: 265-270.
PubMedDirect Link - Schuur, J.H., P. Selzer and J. Gasteiger, 1996. The coding of the three-dimensional structure of molecules by molecular transforms and Its application to structure-spectra correlations and studies of biological activity. J. Chem. Inform. Comput. Sci., 36: 334-344.
CrossRef - Schuur, J. and J. Gasteiger, 1997. Infrared spectra simulation of substituted benzene derivatives on the basis of a 3D structure representation. Anal. Chem., 69: 2398-2398.
CrossRef - Tiziano, T., C. Vincenzo, R. Simona and M. Adriano, 2006. Proposal of a new binding orientation for non-peptide AT1 antagonists: Homology modeling, docking and three dimensional quantitative structure-activity relationship analysis. J. Med. Chem., 49: 4305-4316.
CrossRefPubMedDirect Link