
Mohammad Asif
Department of Pharmacy, GRD (PG) IMT, Dehradun, 248009, Uttarakhand, India
Science International
Year: 2013 | Volume: 1 | Issue: 10 | Page No.: 336-349







ABSTRACT
Background: Quinolones are an important synthetic antimicrobial agents being used over the past three decades. Currently some quinolones are under investigation for the treatment of Mycobacterium tuberculosis and resistant strains of mycobacterium. Results: Their main biological target is the DNA gyrase, a topoisomerase-II encoded by gyrA and gyrB that is essential to maintain the DNA supercoil. The aim of this study is to collect information of the Fluoroquinolones (FQs) which are effective against tuberculosis (TB) and multidrug resistant (MDR-TB), extensively drug resistant-TB (XDR-TB) and latent TB and encourage developing of new QFs which are more effective, less toxic and shorter treatment period. Conclusion: The antitubercular (anti-TB) properties of the quinolone derivatives are as a new class of compounds with potent and selective anti-TB activities. Furthermore, particularly interesting is their activity against multidrug resistance and extensive drug resistance tuberculosis.
PDF Abstract XML References Citation
How to cite this article
Mohammad Asif, 2013. Antimicrobial and Anti-tubercular Activity of Quinolone Analogues. Science International, 1: 336-349.
URL: https://scialert.net/abstract/?doi=sciintl.2013.336.349
URL: https://scialert.net/abstract/?doi=sciintl.2013.336.349
INTRODUCTION
Infectious diseases caused by bacteria have increased dramatically in recent years. Inspite of many significant advances in antibacterial therapy, the widespread use and misuse of antibiotics have caused the emergence of bacterial resistance to antibiotics which is a serious threat to public health. In particular, the emergence of multidrug resistant (MDR) gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant S. aureus (VRSA) and vancomycin-resistant Enterococci (VRE), has become a serious problem in the treatment of bacterial diseases. Therefore, the development of new compounds to deal with resistant bacteria has become one of the most important areas of antibacterial research today. In addition to the development of new and efective antibacterial agents against MDR gram-positive bacteria, recently attention has focused on the treatment of tuberculosis (TB)1. The first quinolone, nalidixic acid (NA), was isolated as a by-product of the synthesis of chloroquine and available for the treatment of urinary tract infections (UTIs). The introduction of fluorinated 4-quinolones, such as ciprofloxacin (CPFX), moxifloxacin (MFLX) and gatifloxacin (GFLX) represents a particularly important therapeutic advance because these agents have broad antimicrobial activity and are effective after oral administration for the treatment of a wide variety of infectious diseases. Relatively few side effects appear to accompany the use of these Fluoroquinolones (FQs) and microbial resistance to their action does not develop rapidly2. Rare and potentially fatal side effects, however, have resulted in the withdrawal from the market of temafloxacin (immune hemolytic anemia), levofloxacin (LVFX) (hepatotoxicity), grepafloxacin (cardiotoxicity) and clinafloxacin (phototoxicity)3.
Tuberculosis (TB) is a common infectious disease and caused by Mycobacterium tuberculosis infection4. The currently available medications show serious side effects like hepatotoxicity [Isoniazid (INH)], damage to auditory nerve [streptomycin (STR)] and thrombocytopenic purpura [rifampicin (RIF)]5. The emergence of MDR-TB has further complicated complicated the therapy6. More frightening is the emergence of extensively drug resistant-TB (XDR-TB) reported in all around the world7. The increased number of MDR strains, is closely associated to the growing global HIV/AIDS pandemic8. The association of TB and HIV infections is so dramatic and patients diagnosed with TB are also HIV-1 positive9. The risk of emergent TB is greater in people living with HIV than those who do not have HIV infection10. TB is the main cause of death of HIV infected people11,12. The immunosuppression linked to HIV infection has also caused the emergence of many opportunistic infections, including disseminated M. avium complex (MAC) infections13,14. The immunosuppression linked to HIV infection has also caused the emergence of many opportunistic infections, including disseminated MAC infections15. The treatment of disseminated MAC infection includes the use of macrolides (clarithromycin or azithromycin) in combination with ethambutol (EBM), or rifabutin (RFB). Centers for Disease Control and Prevention (CDC) notified health care professionals of revised recommendations against the use of rifampicin (RIF) plus pyrazinamide (PZA) for treatment of hospitalization and death from liver injury associated with the combined use of these drugs16.
The aim of this study is to collect information of the FQs which are effective against TB and MDR-TB, XDR-TB and latent TB and encourage developing of new QFs which are more effective, less toxic and shorter treatment period.
CHEMOTHERAPY
The introduction of the combination of streptomycin (STR) and para-aminosalicylic acid (ASA) in therapy and successively, the addition of isoniazid (INH), ethambutol (EMB), rifampicin (RIF) and pyrazinamide (PZA), used in various combinations, resulted in a significant decrease in the mortality17. However, quinolines are classified as second-line drugs, since their use in TB treatment still remains controversial18. On the contrary, they are recommended in managing MDR-TB, due to they have a broad and potent spectrum of activity, giving a better chance of cure and preventing the development and spread of further resistance19. The 4-quinolones-3-carboxylic acid pattern, typical of quinolones, have also been reported to display “non-classical” biological activities, such as antitumor, anxiolitic, anti-ischemic, anti-HIV-1 etc20,21. However, quinolones remain one of the most widely prescribed antibiotics. Quinolones are classified in four generations; The first generation is without fluorine as nalidixic acid (NA), cinoxacin and oxolinic acid (Fig. 1a); The second generation is norfloxacin (NRFX), CPFX, ofloxacin (OFLX), enoxacin and lomefloxacin (Fig. 1b); Third generation is LVFX, sparfloxacin (SPFX) and GFLX (Fig. 1c) and The fourth generation is MFLX and trovafloxacin (TVFX) (Fig. 1d)22. Most FQs are being evaluated as potential anti-TB drugs, also for their potential to shorten TB treatment duration, one of the major strategies for TB control23. WHO also recommends the use of LVFX or MFLX for the treatment of XDR-TB, defined as resistance to INH, RIF, a FQ and a second-line injectable drug, even when OFLX resistance is present24,25.
Fluorine-containing nalidixic acid (NA) derivatives, the FQs, were introduced into clinical practice22. NRFX, the first of a new generation of FQ antibacterials, a fluorine atom at position 6 and a piperazinyl group at position 7 conferred broad and potent antimicrobial activities26,27. Further substitutions of the FQ resulted in the development of CPFX, a widely used antimicrobial agent28. Several modifications of the FQ have been attempted in order to synthesized new derivatives with expanded antimicrobial spectrum, improved pharmacokinetic profiles, decreased selection of resistant mutants, reduced adverse effects and improved efficacy2,29. Last-generation FQs share a broad-spectrum antimicrobial activity against aerobic and anaerobic Gram-positive and Gram-negative bacteria as well as mycobacterium species30,31. Fluoroquinilones (FQs) are recommended and widely used for the treatment of community and nosocomial infections of the respiratory, gastrointestinal and urinary tracts, sexually transmitted diseases, skin and soft tissue infections and chronic osteomyelitis32,33.
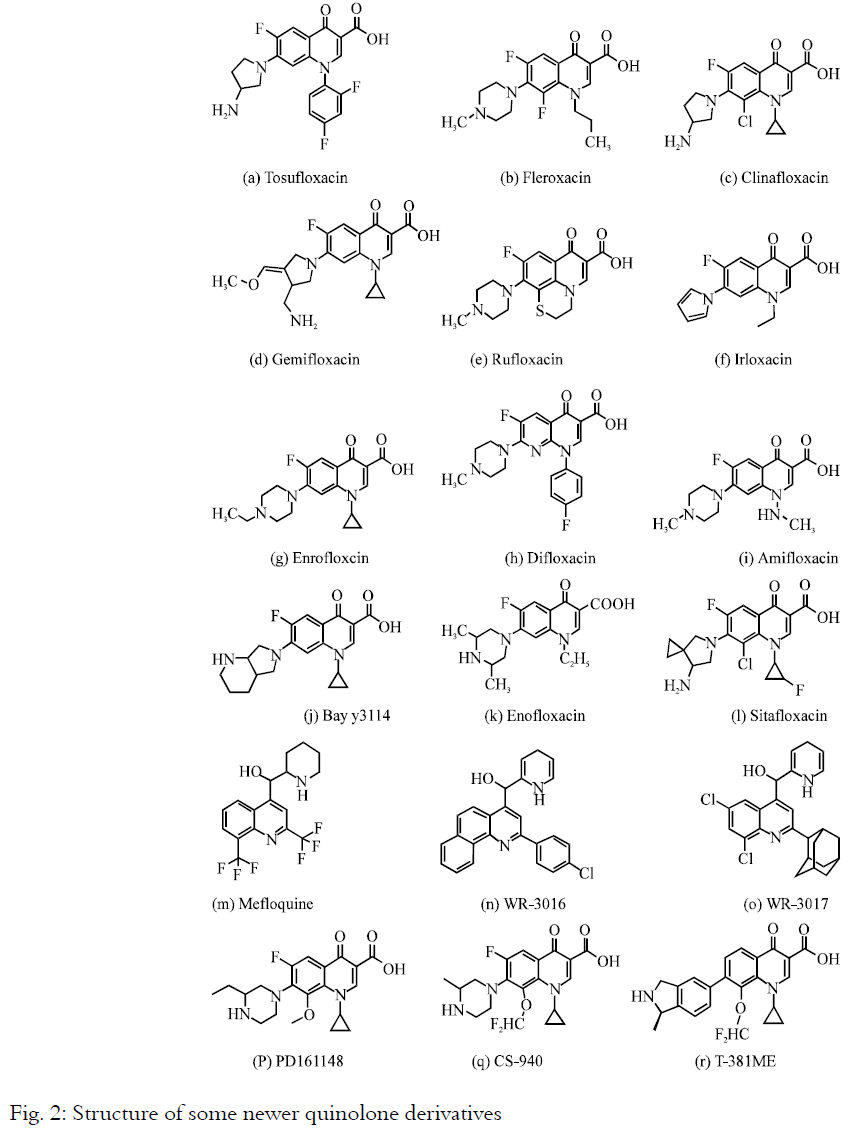
New FQs are in various phases of clinical development, include tosufloxacin, fleroxacin, clinafloxacin, gemifloxacin, rufloxacin, enrofloxacin, difloxacin, amifloxacin, iloxacin, Bay y 3114, temafloxacin, nadifloxacin, grepafloxacin, balofloxacin, pazufloxacin, prulifloxacin, sitafloxacin (STFX), garenoxacin (BMS-284756, T-3811 ME), CFC-222, PD161148, olamufloxacin (HSR-903), WQ-3034, DQ-113, PGE 9262932, PGE 9509924 (Fig. 2)34,35. In particular, MFLX and GFLX are undergoing phase III trials36. Appreciable efficacies of FQs have also been demonstrated against both M. fortuitum infection (particularly when they are included in multidrug regimens)14,37 and M. kansasii and M. xenopi31,38. Many new FQs indicated for the treatment of respiratory tract infections show excellent in vitro activity against MAC isolates39,40. Antimicrobial activity and clinical efficacy of SPFX and LVFX (MIC: 0.5-32 μg mL–1) and GFLX (MIC: 0.25-8 μg mL–1) in MAC infection41,42. FQs can also exert clinical efficacy against mycobacteria when they are administered in combination with other drugs including RIF, INH, PZA, EMB and some aminoglycosides43,.44,45,46,47. The effectiveness of FQs in the treatment of mycobacterial infections may be attributed to the good penetration into infected macrophages where they exert bactericidal activity48,49. Certain drugs, such as RIF, RFB, INH, clofazimine (CLO) and some FQs, strongly or moderately reduced the anti-MAC activity50. The major problem linked with the use of FQs is the increased incidence of FQ-resistant strains of M. tuberculosis (Table 1). Finally, the emergence of MDR-TB strains is related to previous TB treatment of patients with FQs51,52.
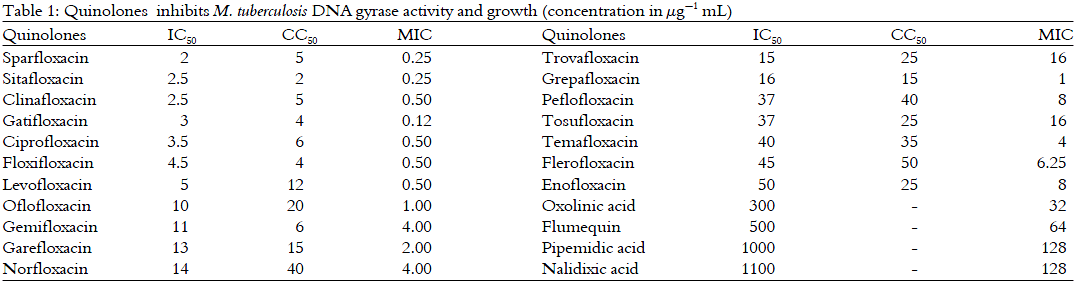 |
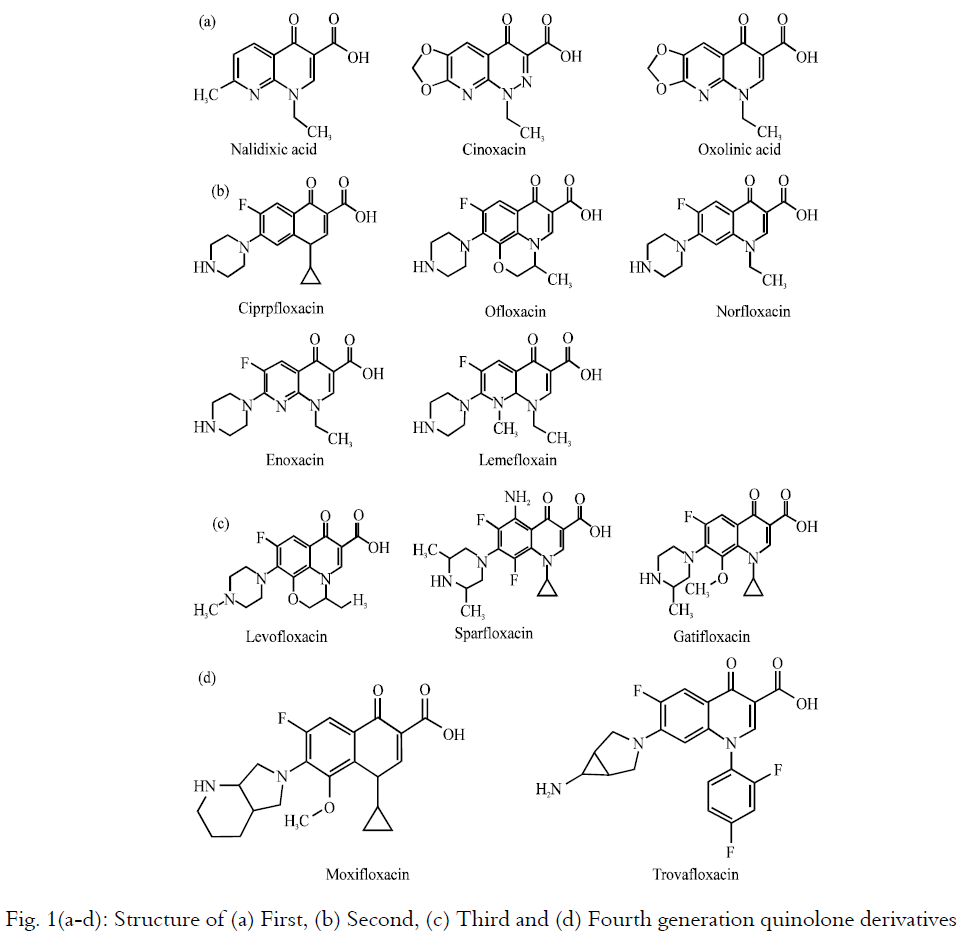 |
MECHANISM OF ACTION OF THE QUINOLONES
The quinolone antibiotics target bacterial DNA gyrase and topoisomerase IV53. For many gram-positive bacteria (such as S. aureus), topoisomerase IV is the primary activity inhibited by the quinolones. In contrast, for many gram-negative bacteria (such as E. coli), DNA gyrase is the primary quinolone target54,55. The individual strands of double-helical DNA must be separated to permit DNA replication or transcription. However, anything that separates the strands results in "overwinding" or excessive positive supercoiling of the DNA in front of the point of separation.
 |
To combat this mechanical obstacle, the bacterial enzyme DNA gyrase is responsible for the continuous introduction of negative supercoils into DNA. This is an ATP-dependent reaction requiring that both strands of the DNA be cut to permit passage of a segment of DNA through the break; the break then is resealed. The DNA gyrase of E. coli is composed of two 105,000-dalton A subunits and two 95,000-dalton B subunits encoded by the gyrA and gyrB genes, respectively. The A subunits which carry out the strand-cutting function of the gyrase are the site of action of the quinolones. The drugs inhibit gyrase-mediated DNA supercoiling at concentrations that correlate well with those required to inhibit bacterial growth (0.1-10 mg mL–1). Mutations of the gene that encodes the A subunit polypeptide can confer resistance to these drugs. Topoisomerase IV also is composed of four subunits encoded by the parC and parE genes in E. coli53,56. Topoisomerase IV separates interlinked (catenated) daughter DNA molecules that are the product of DNA replication. Eukaryotic cells do not contain DNA gyrase. However, they do contain a conceptually and mechanistically similar type II DNA topoisomerase that removes positive supercoils from eukaryotic DNA to prevent its tangling during replication. This enzyme is the target for some antineoplastic agents. Quinolones inhibit eukaryotic type II topoisomerase only at much higher concentrations (100 to 1000 mg mL–1)57. Quinolones exert their anti-TB activity by interfering with two essential ATP-dependent enzymes belonging to the superfamily of topoisomerases IIA: DNA gyrase and topoisomerase IV in bacteria. These enzymes caused different functions: gyrase controls DNA supercoiling and relieves topological stress arising from the translocation of transcription and replication complexes along DNA while topoisomerase IV is a decatenating enzyme that determined interlinked daughter chromosomes following DNA replication53,58. Both enzymes are essential for cell growth and division and are involved in DNA replication, decatenation, recombination and repair59, 60, 61. Gyrase and topoisomerase IV from both Staphylococcus aureus and E. coli have been purified and studied for FQ sensitivity. FQs bind to the DNA/DNAgyrase complex62,63 and preventing the bacterial chromosome from rejoining64. This interferes with DNA repair, replication processes as well as transcription and ultimately leads to bacterial cell death65. In some bacterial species, such as E. coli and Neisseria gonorrhoeae, the primary target is gyrase; in other bacteria, such as S. aureus and Streptococcus pneumonia the primary target is generally topoisomerase IV. Since the two enzymes have different functions, it is likely that bacteria will differ in their response to the quinolones according to which enzyme is the primary target53. For nalidixic and oxolinic acids, end release may come largely from removal of quinolone-gyrase complexes from DNA. For the FQs of the CPFX class, it has been proposed that DNA ends arise from both complex removal and dissociation of gyrase/topoisomerase IV subunits attached to broken DNA. NRFX may be less effective in separating the gyrase subunits. Newer FQs, such as SPFX and DU6859a which carry C-8 substituents, seem to attack gyrase more effectively66,67.
RESISTANCE TO QUINOLONES
Genome studies have shown that there is no evidence of the topoisomerase IV gene homologs in the genome of M. tuberculosis68, suggesting that DNA gyrase is the sole type II topoisomerase and is likely to be the unique target of quinolones in M. tuberculosis69. Most mutations conferring bacterial resistance to quinolones occur in a short discrete segment termed the Quinolone Resistance-Determining Region (QRDR) of the DNA gyrase gyrA gene59,70. Diversely, in M. tuberculosis mutations in two short QRDR have been associated with FQ resistance71 is GyrA subunit (QRDR-A) and GyrB (QRDR-B)72,73. The most common mutation in FQ-resistant M. tuberculosis isolates74,75. A similar mutation has been observed in the topoisomerase IV of bacteria23,76. The mechanism of resistance to FQs in M. tuberculosis was studied by selecting spontaneous FQ-resistant mutants from susceptible strains, H37Rv and H37Ra77,78. The genes that encode the DNA gyrase of M. tuberculosis H37Rv have been cloned and sequenced and mutations associated with quinolone resistance have been found in the gyrA gene from clinical isolates79,80. Studies on M. tuberculosis have revealed that single missense mutations in gyrA are associated with low-level quinolone resistance and that bacteria with high-level resistance generally have two missense mutations in gyrA or one mutation in gyrA and one in gyrB67. Different resistance mechanisms, often interdependent may explain various degrees of resistance and also may account for the stepwise selection of highly FQ-resistant strains81,82. Furthermore, target gene mutations may differ from one geographic region to another83. Genes encoding drug efflux transporters have been isolated from several mycobacterial species. These proteins transport tetracycline, FQs, aminoglycosides and other compounds. The balance between the drug transport into the cell and drug efflux is not yet clearly understood and further studies are required in mycobacterium84.
PHARMACOKINETIC AND PHARMACODYNAMIC PARAMETERS
The pharmacokinetic and pharmacodynamic parameters of antimicrobial agents are important in preventing the selection and spread of resistant strains and have led to description of the mutation-prevention concentration which is the lowest concentration of antimicrobial that prevents selection of resistant bacteria from high bacterial inocula. β-Lactams are time-dependent agents without significant postantibiotic effects, resulting in bacterial eradication when unbound serum concentrations exceed MICs of these agents against infecting pathogens for more than 40-50% of the dosing interval. By contrast, FQs are concentration-dependent agents, resulting in bacterial eradication when unbound serum area-under-the-curve-to-MIC ratios exceed 25-30. These observations are now being used to assess the roles of current agents, develop new formulations and assess potency of new antimicrobials. The FQs can be administered orally, with good absorption not affected by food intake. New FQs reach relevant concentration into alveolar macrophages, bronchial mucosa, epithelial lining fluid and saliva. Specifically, grepafloxacin, MFLX and SPFX display superior penetration into alveolar macrophages and epithelial lining fluid in comparison to CPFX. Tissue and fluid concentrations often exceed serum concentrations. All of the new FQs exhibit longer plasma half-lives compared to CPFX, like STFX and SPFX. Clinafloxacin, GFLX, LVFX and STFX are excreted > 50 % in the urine as the parent compound, indicating that these agents primarily undergo renal excretion. Conversely, gemifloxacin, grepafloxacin, MFLX, SPFX and TVFX are all cleared predominantly by nonrenal ways. The most common adverse effects associated with the use of FQs are gastrointestinal disturbances, nervous system complaints (dizziness, headache) and allergic reactions (skin rashes and pruritus)85,86,87. The use of several FQs have been severely restricted because of advers effects; clinafloxacin causing phototoxicity and hypoglycaemia88, SPFX causing phototoxicity89 and TVFX causing hepatotoxicity90. Grepafloxacin has been withdrawn from the market due to prolongation of the QTc interval. Other FQs such as GFLX, gemifloxacin, LVFX and MFLX have toleraility issues comparable to CPFX. Drug interactions are limited and are infrequent between FQs and other antit-TB drugs91, however FQ absorption may be reduced when coadministered with antacids containing multivalent cations92,93. The physicochemical properties of quinolones (e.g., relative hydrophobicity, charge or molecular mass) are important factors for bacterial cell penetration and splay a different role in Gram-negative and Gram-positive bacteria. Increasing molecular mass and bulkiness of substituents at C-7 position hinder penetration of quinolones into Gram-negative bacteria through the porin channels, although hydrophobic molecules appear to enter via the lipopolysaccharide or across the lipid bilayer94. Gram-positive bacteria do not possess an outer membrane, therefore lacking outer membrane proteins and lipopolysaccharide. Intracellular accumulation observed in Gram-positive bacteria (e.g., S. aureus) is thought to take place by simple diffusion across the cytoplasmic membrane95,96. The unique cell wall structure of mycobacteria is rich in long-chain fatty acids such as C60 to C90 mycolic acids97,98. Mycolic acids are covalently linked to the peptidoglycan-associated polysaccharide arabinogalactan. Moreover, mycobacterial porins, the water-filled channel proteins which form the hydrophilic diffusion pathways are sparse99. A major porin of M. smegmatis, MspA, forms a tetrameric complex with a single central pore but the density of this protein is 50-fold lower than that of porins of gram negative bacteria100. Thus, the mycobacterial cell wall functions as an even more efficient protective barrier than the outer membrane of gram-negative bacteria and limits the access of drug molecules to their cellular targets.
ANTIBACTERIAL SPECTRUM
The FQs are potent bactericidal agents against E. coli and various species of Salmonella, Shigella, Enterobacter, Campylobacter and Neisseria. CPFX is more active than NRFX against P. aeruginosa. FQs also have good activity against staphylococci but not against methicillin-resistant strains. Activity against streptococci is limited to a subset of the quinolones, including LVFX, GFLX and MFLX101. Several intracellular bacteria are inhibited by FQs at concentrations that can be achieved in plasma; these include species of Chlamydia, Mycoplasma, Legionella, Brucella and Mycobacterium102,103. CPFX, OFLX and pefloxacin have MIC90 values from 0.5-3 mg mL–1 for M. fortuitum, M. kansasii and M. tuberculosis; OFLX and pefloxacin are active in animal models of leprosy56. Several of the new FQs have activity against anaerobic bacteria, including garenoxacin and gemifloxacin104. Resistance to quinolones may develop during therapy via mutations in the bacterial chromosomal genes encoding DNA gyrase or topoisomerase IV or by active transport of the drug out of the bacteria105. No quinolone-modifying or-inactivating activities have been identified in bacteria106. Resistance has increased after the introduction of FQs, especially in Pseudomonas and staphylococci107. Increasing FQ resistance also is being observed in C. jejuni, Salmonella, N. gonorrhoeae and S. pneumoniae108.
THERAPEUTIC USES
Urinary tract infections: Nalidixic acid (NA) is useful only for UTIs caused by microbs. The FQs are significantly more potent and have a much broader spectrum of antimicrobial activity. NRFX is use for UTIs. Comparative clinical trials indicate that the FQs are more efficacious than trimethoprim-sulfamethoxazole for the treatment of UTIs109.
Prostatitis: The NRFX, CPFX and OFLX all have been effective in uncontrolled trials for the treatment of prostatitis caused by sensitive bacteria. FQs appear to be effective in patients not responding to trimethoprim-sulfamethoxazole109.
Sexually transmitted diseases: The quinolones are contraindicated in pregnancy. FQs lack activity for Treponema pallidum but have activity in vitro against N. gonorrhoeae, C. trachomatis and H. ducreyi. For chlamydial urethritis/cervicitis, a course of OFLX or SPFX is an alternative to a treatment with doxycycline or a single dose of azithromycin; other available quinolones have not been reliably effective. A single oral dose of a FQ such as OFLX or CPFX is effective treatment for sensitive strains of N. gonorrhoeae but increasing resistance to FQs has led to ceftriaxone being the first-line agent for this infection. Pelvic inflammatory disease has been treated effectively with a course of OFLX combined with an antibiotic with activity against anaerobes (clindamycin or metronidazole)110. Chancroid (infection by H. ducreyi) can be treated with CPFX.
Gastrointestinal and abdominal infections: For traveler’s diarrhea (caused by enterotoxigenic E. coli), the quinolones are equal to trimethoprim-sulfamethoxazole in effectiveness, reducing the duration of loose stools by 1-3 days111. NRFX, CPFX and OFLX have been effective in the treatment of patients with shigellosis112. NRFX is superior to tetracyclines in decreasing the duration of diarrhea in cholera. CPFX and OFLX treatment cures most patients with enteric fever caused by S. typhi, as well as bacteremic nontyphoidal infections in AIDS patients and it clears chronic fecal carriage. Shigellosis is treated effectively with either CPFX or azithromycin113. The ability of the quinolones to induce the Shiga toxin stx2 gene (the cause of the hemolytic-uremic syndrome) in E. coli suggests that the quinolones should not be used for Shiga toxin-producing E. coli114. CPFX and OFLX have been less effective in treating episodes of peritonitis occurring in patients on chronic ambulatory peritoneal dialysis likely owing to the higher MICs for these drugs for the coagulase-negative staphylococci that are a common cause of peritonitis in this setting.
Respiratory tract infections: The major limitation to the use of quinolones for the treatment of community-acquired pneumonia and bronchitis had been the poor in vitro activity of CPFX, OFLX and NRFX against S. pneumoniae and anaerobic bacteria. However, many of the newer FQs, including GFLX and MFLX, have excellent activity against S. pneumoniae. Initial clinical experience with some of these newer quinolones shows comparable efficacy to β-lactam antibiotics115. The FQs have in vitro activity against the rest of the commonly recognized respiratory pathogens, including H. influenzae, Moraxella catarrhalis, S. aureus, M. pneumoniae, Chlamydia pneumoniae and Legionella pneumophila. Either a FQ (CPFX or LVFX) or azithromycin is the antibiotic of choice for L. pneumophila116. FQs have been very effective at eradicating both H. influenzae and M. catarrhalis from sputum. Mild to moderate respiratory exacerbations owing to P. aeruginosa in patients with cystic fibrosis have responded to oral FQ therapy. FQs used as single agents for treatment of community-acquired pneumonia56 and decreasing susceptibility of S. pneumoniae to Fqs117,118.
Bone, joint and soft tissue infections: The treatment of chronic osteomyelitis requires prolonged antimicrobial therapy with agents active against S. aureus and gram-negative rods119. Bone and joint infections may require treatment for 4 to 6 weeks or more. Dosage should be reduced for patients with severely impaired renal function. CPFX should not be given to children or pregnant women. Failures have been associated with the development of resistance in S. aureus, P. aeruginosa and Serratia marcescens. In diabetic foot infections which are commonly caused by a mixture of bacteria including gram-negative rods, anaerobes, streptococci and staphylococci, the FQs in combination with an agent with antianaerobic activity are a reasonable choice. CPFX as sole therapy is effective in 50% of diabetic foot infections107.
Other infections: The CPFX received wide usage for the prophylaxis of anthrax and has been shown to be effective for the treatment of tularemia120,121. The quinolones may be used as part of multiple-drug regimens for the treatment of MDR-TB and for the treatment of atypical TB infections as well as MAC infections in AIDS. In neutropenic cancer patients with fever, the combination of a quinolone with an aminoglycoside is comparable to β-lactam-aminoglycoside combinations; quinolones are less effective when used alone. Quinolones, when used as prophylaxis in neutropenic patients, have decreased the incidence of gram-negative rod bacteremias. CPFX plus amoxicillin-clavulanate has been shown recently to be effective as an oral empirical therapy for fever in low-risk patients with granulocytopenia secondary to cancer chemotherapy.
Adverse effects: Quinolones and FQs generally are well tolerated122. The most common adverse reactions involve the GI tract, with 3-17% of patients reporting mostly mild nausea, vomiting and/or abdominal discomfort. Diarrhea and antibiotic-associated colitis have been unusual. CNS side effects, mild headache and dizziness are seen in 0.9-11% of patients. Rarely, hallucinations, delirium and seizures have occurred, predominantly in patients who also were receiving theophylline or a nonsteroidal antiinflammatory drug (NSAIDs). CPFX and pefloxacin inhibit the metabolism of theophylline and toxicity from elevated concentrations of the methylxanthine may occur123. NSAIDs may augment displacement of g-aminobutyric acid (GABA) from its receptors by the quinolones124. Rashes, including photosensitivity reactions, also can occur. Achilles tendon rupture or tendinitis has occurred rarely. Renal disease, hemodialysis and steroid use may be predisposing factors122. Traditionally, the use of quinolones in children has been contraindicated for this reason. However, children with cystic fibrosis given CPFX, NRFX and NA have had few and reversible, joint symptoms125. Therefore, in some cases the benefits may outweigh the risks of quinolone therapy in children. Leukopenia, eosinophilia and mild elevations in serum transaminases occur rarely. QTc interval (QT interval corrected for heart rate) prolongation has been observed with SPFX and to a lesser extent with GFLX and MFLX. Quinolones probably should be used only with caution in patients on class III (amiodarone) and class IA (quinidine, procainamide) antiarrhythmics.
ABSORPTION, FATE and EXCRETION
The quinolones are well absorbed after oral administration and are distributed widely in body tissues. Peak serum levels of the FQs are obtained within 1-3 h of an oral dose of 400 mg, with peak levels ranging from 1.1 for SPFX to 6.4 mg mL–1 for LVFX. Relatively low serum levels are reached with NRFX and limit its usefulness to the treatment of UTIs. Food does not impair oral absorption but may delay the time to peak serum concentrations. Oral doses in adults are 200-400 mg every 12 h for OFLX, 400 mg every 12 h for NRFX and pefloxacin and 250-750 mg every 12 h for CPFX. Bioavailability of the FQs is greater than 50% for all agents and greater than 95% for several. The serum half-life ranges from 3-5 h for NRFX and CPFX to 20 h for SPFX. The volume of distribution of quinolones is high, with concentrations of quinolones in urine, kidney, lung and prostate tissue, stool, bile and macrophages and neutrophils higher than serum levels. Quinolone concentrations in cerebrospinal fluid, bone and prostatic fluid are lower than in serum. Pefloxacin and OFLX levels in ascites fluid are close to serum levels and CPFX, OFLX and pefloxacin have been detected in human breast milk. Most quinolones are cleared predominantly by the kidney and dosages must be adjusted for renal failure. Exceptions are pefloxacin and MFLX which are metabolized predominantly by the liver and should not be used in patients with hepatic failure. None of the agents is removed efficiently by peritoneal dialysis or hemodialysis53,56.
DISCUSSION
Ever since the discovery of NA in the early 1960s, DNA gyrase, a prokaryotic-specific type II topoisomerase enzyme, has attracted considerable attention from the pharmaceutical industry126. It belongs to a superfamily of ATPases known as GHKL (gyrase, Hsp90, histidine, kinase, MutL) which consists of several important targets for infectious treatments. Although similar to eukaryotic topoisomerase-2, DNA gyrase has many differences such that certain antibiotics act specifically against it127. Bacterial DNA gyrase is involved in the vital processes of DNA replication, transcription and recombination128. The structure activity relationships of compounds based on NA have led to a large group of synthetic antibacterial agents known collectively as quinolones. The 4-quinolones such as CPFX, OFLX, lomefloxacin, enoxacin are established synthetic antibacterial agents as DNA gyrase inhibitors129. The 2-quinolones, also called carbostyrils or 1-aza coumarins are isosteric to coumarins and isomeric to 4-quinolones and could become a potential candidate for antibacterial activity130. Reports are available on DNA gyrase inhibition by quinolones131,132. Quinolones are known to inhibit the DNA breakage-reunion cycle by binding to the subunit A and by blocking the gyrase-DNA complex, whereas the latter act on subunit B133,134.
CONCLUSION
Many human illnesses are caused by infections with microbes like viruses or bacteria or fungi. Amongst those various illnesses, certain tubercular, bacterial, viral and fungal infections are more common because of their tendency to develop new strains under any circumstances and developing resistance against the available drugs. This stimulated the scientists for development of novel molecules to combat these illnesses. Infectious microbial disease remains a pressing problem worldwide, because microbes have resisted prophylaxis or therapy longer than any other form of life. In conclusion, it can confirm that in general quinolones are particularly adapted to be used as antitubercular agents. Finally, the selectivity and the consistent ability to reduce the onset of cross resistance, probably due to a different mechanism of action toward quinolones, lead them to be good candidates for further development.
REFERENCES
- Dogruer, D.S., T. Onkol, S. Ozkan, S. Ozgen and M.F. Sahin, 2008. Synthesis and antimicrobial activity of some 3 (2H)-pyridazinone and 1(2H)-phthalazinone derivatives. Turk. J. Chem., 32: 469-479.
Direct Link - Grosset, J., L. Zunic and C. Morcrette, 2002. World epidemiology of tuberculosis and resistance against anti-tuberculosis drugs. Ann. Med. Int., 153: 107-112.
PubMed - Ginsburg, A.S., N. Hooper, N. Parrish, K.E. Dooley and S.E. Dorman, 2003. Fluoroquinolone resistance in patients with newly diagnosed tuberculosis. Clin. Infect. Dis., 37: 1448-1452.
PubMed - Getahun, H., C. Gunneberg, R. Granich and P. Nunn, 2010. HIV infection-associated tuberculosis: The epidemiology and the response. Clin. Infect. Dis., 50: S201-S207.
CrossRef - Hart, C.A., N.J. Beeching and B.I. Duerden, 1996. Tuberculosis into the next century: Proceedings of a symposium held on 4 February 1995 at the Liverpool School of Tropical Medicine. J. Med. Microbiol., 44: 1-34.
CrossRefDirect Link - Tomioka, H., K. Sato, H. Kajitani, T. Akaki and S. Shishido, 2000. Comparative antimicrobial activities of the newly synthesized quinolone WQ-3034, levofloxacin, sparfloxacin and ciprofloxacin against Mycobacterium tuberculosis and Mycobacterium avium complex. Antimicrob. Agents Chemother., 44: 283-286.
CrossRef - Tomioka, H., 2000. Prospects for development of new antimycobacterial drugs. J. Infect. Chemother., 6: 8-20.
CrossRef - Migliori, G.B., C. Lange, E. Girardi, R. Centis and G. Besozzi et al., 2008. Fluoroquinolones: Are they essential to treat multidrug-resistant tuberculosis? Eur. Respir. J., 31: 904-905.
CrossRef - Mugnaini, C., S. Pasquini and F. Corelli, 2009. The 4-quinolone-3-carboxylic acid motif as a multivalent scaffold in medicinal chemistry. Curr. Med. Chem., 16: 1746-1767.
CrossRef - Ahmed, A. and M. Daneshtalab, 2012. Nonclassical biological activities of quinolone derivatives. J. Pharm. Pharm. Sci., 15: 52-72.
PubMed - De Souza, M.V.N., T.R. Vasconcelos, M.V. Almeida and S.H. Cardoso, 2006. Fluoroquinolones: An important class of antibiotics against tuberculosis. Curr. Med. Chem., 13: 455-463.
Direct Link - Van den Boogaard, J., G.S. Kibiki, E.R. Kisanga, M.J. Boeree and R.E. Aarnoutse, 2009. New drugs against tuberculosis: Problems, progress and evaluation of agents in clinical development. Antimicrob. Agents Chemother., 53: 849-862.
CrossRef - Devasia, R.A., A. Blackman, C. May, S. Eden and T. Smith et al., 2009. Fluoroquinolone resistance in Mycobacterium tuberculosis: An assessment of MGIT 960, MODS and nitrate reductase assay and fluoroquinolone cross-resistance. J. Antimicrob. Chemother., 63: 1173-1178.
CrossRef - Appelbaum, P.C. and P.A. Hunter, 2000. The fluoroquinolone antibacterials: Past, present and future perspectives. Int. J. Antimicrob. Agents, 16: 5-15.
CrossRef - Sharma, A.K., R. Khosla, A.K. Kela and V.L. Mehta, 1994. Fluoroquinolones: Antimicrobial agents of the 90's. Indian J. Pharmacol., 26: 249-261.
Direct Link - Zhanel, G.G., K. Ennis, L. Vercaigne, A. Walkty and A.S. Gin et al., 2002. A critical review of the fluoroquinolones: Focus on respiratory infections. Drugs, 62: 13-59.
PubMed - Domagala, J.M., 1994. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother., 33: 685-706.
CrossRef - Gillespie, S.H., I. Morrissey and D. Everett, 2001. A comparison of the bactericidal activity of quinolone antibiotics in a Mycobacterium fortuitum model. J. Med. Microbiol., 50: 565-570.
PubMed - Jones, M.E., I.A. Critchley, J.A. Karlowsky, R.S. Blosser-Middleton, F.J. Schmitz, C. Thornsberry and D.F. Sahm, 2002. In vitro activities of novel nonfluorinated quinolones PGE 9262932 and PGE 9509924 against clinical isolates of Staphylococcus aureus and Streptococcus pneumoniae with defined mutations in DNA gyrase and topoisomerase IV. Antimicrob. Agents Chemother., 46: 1651-1657.
CrossRef - Richeldi, L., M. Covi, G. Ferrara, F. Franco and P. Vailati et al., 2002. Clinical use of levofloxacin in the long term treatment of drug resistant tuberculosis. Monaldi Archiv. Chest Dis., 57: 39-43.
PubMedDirect Link - Rodriguez, J.C., L. Cebrian, M. Lopez, M. Ruiz and G. Royo, 2005. Usefulness of various antibiotics against Mycobacterium avium-intracellulare, measured by their mutant prevention concentration. Int. J. Antimicrob. Agents, 25: 221-225.
CrossRef - Woods, G.L., N. Williams-Bouyer, R.J. Wallace Jr., B.A. Brown-Elliott and F.G. Witebsky et al., 2003. Multisite reproducibility of results obtained by two broth dilution methods for susceptibility testing of Mycobacterium avium complex. J. Clin. Microbiol., 41: 627-631.
CrossRef - Horiguchi, T., R. Kondo, J. Miyazaki, M. Shiga, M. Sugiyama, M. Handa and E. Munekata, 2004. Antibacterial activity and clinical efficacy of sparfloxacin in Mycobacterium avium-intracellulare complex infection. J. Int. Med. Res., 32: 530-539.
CrossRef - Kobashi, Y., K. Yoshida, N. Miyashita, Y. Niki and M. Oka, 2006. Relationship between clinical efficacy of treatment of pulmonary Mycobacterium avium complex disease and drug-sensitivity testing of Mycobacterium avium complex isolates. J. Infect. Chemother., 12: 195-202.
CrossRef - Tomioka, H., C. Sano, K. Sato and T. Shimizu, 2002. Antimicrobial activities of clarithromycin, gatifloxacin and sitafloxacin, in combination with various antimycobacterial drugs against extracellular and intramacrophage Mycobacterium avium complex. Int. J. Antimicrob. Agents, 19: 139-145.
CrossRef - Fattorini, L., D. Tan, E. Iona, M. Mattei and F. Giannoni et al., 2003. Activities of moxifloxacin alone and in combination with other antimicrobial agents against multidrug-resistant Mycobacterium tuberculosis infection in BALB/c mice. Antimicrob. Agents Chemother., 47: 360-362.
CrossRef - Sano, K., H. Tomioka, K. Sato, C. Sano, H. Kawauchi, S. Cai and T. Shimizu, 2004. Interaction of antimycobacterial drugs with the anti-Mycobacterium avium complex effects of antimicrobial effectors, reactive oxygen intermediates, reactive nitrogen intermediates and free fatty acids produced by macrophages. Antimicrob. Agents Chemother., 48: 2132-2139.
CrossRefDirect Link - Cambau, E. and V. Jarlier, 1996. Resistance to quinolones in mycobacteria. Res. Microbiol., 147: 52-59.
CrossRefPubMedDirect Link - Ginsburg, A.S., J.H. Grosset and W.R. Bishai, 2003. Fluoroquinolones, tuberculosis and resistance. Lancet Infect. Dis., 3: 432-442.
CrossRefPubMedDirect Link - Drlica, K. and X. Zhao, 1997. DNA gyrase, topoisomerase IV and the 4-quinolones. Microbiol. Mol. Biol. Rev., 61: 377-392.
PubMedDirect Link - Hooper, D.C., 2000. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis., 31: S24-S28.
PubMed - Alovero, F.L., X.S. Pan, J.E. Morris, R.H. Manzo and L.M. Fisher, 2000. Engineering the specificity of antibacterial fluoroquinolones: benzenesulfonamide modifications at C-7 of ciprofloxacin change its primary target in Streptococcus pneumoniae from topoisomerase IV to gyrase. Antimicrob. Agents Chemother., 44: 320-325.
CrossRefPubMed - Anquetin, G., J. Greiner, N. Mahmoudi, M. Santillana-Hayat and R. Gozalbes et al., 2006. Design, synthesis and activity against Toxoplasma gondii, Plasmodium spp. and Mycobacterium tuberculosis of new 6-fluoroquinolones. Eur. J. Med. Chem., 41: 1478-1493.
CrossRefPubMedDirect Link - Barnard, F.M. and A. Maxwell, 2001. Interaction between DNA gyrase and quinolones: effects of alanine mutations at GyrA subunit residues Ser83 and Asp87. Antimicrob. Agents Chemother., 45: 1994-2000.
CrossRefPubMedDirect Link - Champoux, J.J., 2001. DNA topoisomerases: Structure, function and mechanism. Annu. Rev. Biochem., 70: 369-413.
CrossRefPubMedDirect Link - Mitscher, L.A., 2005. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev., 105: 559-592.
CrossRefPubMedDirect Link - Brighty, K.E. and T.D. Gootz, 1997. The chemistry and biological profile of trovafloxacin. J. Antimicrob. Chemother., 39: 1-14.
PubMedDirect Link - Yoshida, H., M. Nakamura, M. Bogaki, H. Ito, T. Kojima, H. Hattori and S. Nakamura, 1993. Mechanism of action of quinolones against Escherichia coli DNA gyrase. Antimicrob. Agents Chemother., 37: 839-845.
CrossRef - Pestova, E., J.J. Millichap, G.A. Noskin and L.R. Peterson, 2000. Intracellular targets of moxifloxacin: A comparison with other fluoroquinolones. J. Antimicrob. Chemother., 45: 583-590.
CrossRef - Fukuda, H. and K. Hiramatsu, 1999. Primary targets of fluoroquinolones in Streptococcus pneumoniae. Antimicrob. Agents Chemother., 43: 410-412.
PubMedDirect Link - Cole, S.T., R. Brosch, J. Parkhill, T. Garnier and C. Churcher et al., 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature, 393: 537-544.
CrossRefDirect Link - Aubry, A., X.S. Pan, L.M. Fisher, V. Jarlier and E. Cambau, 2004. Mycobacterium tuberculosis DNA gyrase: Interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother., 48: 1281-1288.
CrossRef - Sun, Z., J. Zhang, X. Zhang, S. Wang, Y. Zhang and C. Li, 2008. Comparison of gyrA gene mutations between laboratory-selected ofloxacin-resistant Mycobacterium tuberculosis strains and clinical isolates. Int. J. Antimicrob. Agents, 31: 115-121.
CrossRefDirect Link - Piton, J., S. Petrella, M. Delarue, G. Andre-Leroux, V. Jarlier, A. Aubry and C. Mayer, 2010. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS One, Vol. 5.
CrossRef - Veziris, N., C. Martin, F. Brossier, F. Bonnaud, F. Denis and A. Aubry, 2007. Treatment failure in a case of extensively drug-resistant tuberculosis associated with selection of a GyrB mutant causing fluoroquinolone resistance. Eur. J. Clin. Microbiol. Infect. Dis., 26: 423-425.
CrossRefDirect Link - Xu, C., B.N. Kreiswirth, S. Sreevatsan, J.M. Musser and K. Drlica, 1996. Fluoroquinolone resistance associated with specific gyrase mutations in clinical isolates of multidrug-resistant Mycobacterium tuberculosis. J. Infect. Dis., 174: 1127-1130.
CrossRefDirect Link - Perlman, D.C., W.M. El Sadr, L.B. Heifets, E.T. Nelson and J.P. Matts et al., 1997. Susceptibility to levofloxacin of Mycobacterium tuberculosis isolates from patients with HIV-related tuberculosis and characterization of a strain with levofloxacin monoresistance. Aids, 11: 1473-1478.
Direct Link - Ruiz, J., 2003. Mechanisms of resistance to quinolones: Target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother., 51: 1109-1117.
CrossRefPubMedDirect Link - Kocagoz, T., C.J. Hackbarth, I. Unsal, E.Y. Rosenberg, H. Nikaido and H.F. Chambers, 1996. Gyrase mutations in laboratory-selected, fluoroquinolone-resistant mutants of Mycobacterium tuberculosis H37Ra. Antimicrob. Agents Chemother., 40: 1768-1774.
Direct Link - Foroumadi, A., M. Oboudiat, S. Emami, A. Karimollah, L. Saghaee, M.H. Moshafi and A. Shafiee, 2006. Synthesis and antibacterial activity of N-[2-[5-(methylthio) thiophen-2-yl]-2-oxoethyl] and N-[2-[5-(methylthio) thiophen-2-yl]-2-(oxyimino) ethyl] piperazinylquinolone derivatives. Bioorg. Med. Chem., 14: 3421-3427.
CrossRefDirect Link - Takiff, H.E., L. Salazar, C. Guerrero, W. Philipp and W.M. Huang et al., 1994. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob. Agents Chemother., 38: 773-780.
CrossRefDirect Link - Cambaup, E., W. Sougakoff, M. Besson, C. Truffot-Pernot, J. Grosset and V. Jarlier, 1994. Selection of a gyrA mutant of Mycobacterium tuberculosis resistant to fluoroquinolones during treatment with ofloxacin. J. Infect. Dis., 170: 479-483.
CrossRefDirect Link - Banerjee, S.K., K. Bhatt, S. Rana, P. Misra and P.K. Chakraborti, 1996. Involvement of an efflux system in mediating high level of fluoroquinolone resistance in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun., 226: 362-368.
CrossRefDirect Link - Acar, J.F. and F.W. Goldstein, 1997. Trends in bacterial resistance to fluoroquinolones. Clin. Infect. Dis., 24: S67-S73.
CrossRefDirect Link - Lee, A.S., L.L. Tang, I.H Lim and S.Y. Wong, 2002. Characterization of pyrazinamide and ofloxacin resistance among drug-resistant Mycobacterium tuberculosis isolates from Singapore. Int. J. Infect. Dis., 6: 48-51.
CrossRefDirect Link - Rossi, E.D., J.A Ainsa and G. Riccardi, 2006. Role of mycobacterial efflux transporters in drug resistance: An unresolved question. FEMS Microbiol. Rev., 30: 36-52.
CrossRefDirect Link - Habib, M.P., L.O. Gentry, G. Rodriguez-Gomez, W. Morowitz and E. Polak et al., 1998. Multicenter, randomized study comparing efficacy and safety of oral levofloxacin and cefaclor in treatment of acute bacterial exacerbations of chronic bronchitis. Infect. Dis. Clin. Pract., 7: 101-109.
Direct Link - Lucena, M.I., R.J. Andrade, L. Rodrigo, J. Salmeron and A. Alvarez et al., 2000. Trovafloxacin-induced acute hepatitis. Clin. Infect. Dis., 30: 400-401.
CrossRefDirect Link - Ridzon, R., J. Meador, R. Maxwell, K. Higgins, P. Weismuller and I.M. Onorato, 1997. Asymptomatic hepatitis in persons who received alternative preventive therapy with pyrazinamide and ofloxacin. Clin. Infect. Dis., 24: 1264-1265.
CrossRefDirect Link - Yew, W.W., 2002. Clinically significant interactions with drugs used in the treatment of tuberculosis. Drug Safety, 25: 111-133.
CrossRefDirect Link - Dalhoff, A. and F.J. Schmitz, 2003. In vitro antibacterial activity and pharmacodynamics of new quinolones. Eur. J. Clin. Microbiol. Infect. Dis., 22: 203-221.
CrossRefDirect Link - McCaffrey, C., A. Bertasso, J. Pace and N.H. Georgopapadakou, 1992. Quinolone accumulation in Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus. Antimicrob. Agents Chemother., 36: 1601-1605.
CrossRefDirect Link - Piddock, L.J., M.C Hall and R. Wise, 1990. Mechanism of action of lomefloxacin. Antimicrob. Agents Chemother., 34: 1088-1093.
CrossRefDirect Link - Brennan, P.J. and H. Nikaido, 1995. The envelope of mycobacteria. Annu. Rev. Biochem., 64: 29-63.
CrossRefDirect Link - Trias, J. and R. Benz, 1994. Permeability of the cell wall of Mycobacterium smegmatis. Mol. Microbiol., 14: 283-290.
CrossRefDirect Link - Engelhardt, H., C. Heinz and M. Niederweis, 2002. A tetrameric porin limits the cell wall permeability of Mycobacterium smegmatis. J. Biol. Chem., 277: 37567-37572.
CrossRefDirect Link - Leysen, D.C., A. Haemers and S.R. Pattyn, 1989. Mycobacteria and the new quinolones. Antimicrob. Agents Chemother., 33: 1-5.
PubMed - Alangaden, G.J. and S.A. Lerner, 1997. The clinical use of fluoroquinolones for the treatment of mycobacterial diseases. Clin. Infect. Dis., 25: 1213-1221.
CrossRefPubMedDirect Link - Anonymous, 2000. Gatifloxacin and moxifloxacin: Two new fluoroquinolones. Med. Lett. Drugs Ther., 42: 15-17.
PubMedDirect Link - Oethinger, M., W.V. Kern, A.S. Jellen-Ritter, L.M. McMurry and S.B. Levy, 2000. Ineffectiveness of topoisomerase mutations in mediating clinically significant fluoroquinolone resistance in Escherichia coli in the absence of the AcrAB efflux pump. Antimicrob. Agents Chemother., 44: 10-13.
CrossRefPubMedDirect Link - Gold, H.S. and R.C. Moellering Jr., 1996. Antimicrobial-drug resistance. N. Engl. J. Med., 335: 1445-1453.
CrossRefDirect Link - Peterson, L.R., M. Postelnick, T.L. Pozdol, B. Reisberg and G.A. Noskin, 1998. Management of fluoroquinolone resistance in Pseudomonas aeruginosa-Outcome of monitored use in a referral hospital. Int. J. Antimicrob. Agents, 10: 207-214.
CrossRefPubMedDirect Link - Thornsberry, C., P. Ogilvie, J. Kahn and Y. Mauriz, 1997. Surveillance of antimicrobial resistance in Streptococcus pneumonia, Haemophilus influenza and Moraxella catarrhalis in the United States in 1996-1997 respiratory season. Diagn. Microbiol. Infect. Dis., 29: 249-257.
CrossRefDirect Link - Hooper, D.C. and J.S. Wolfson, 1991. Fluoroquinolone antimicrobial agents. N. Engl. J. Med., 324: 384-394.
CrossRefPubMedDirect Link - Anonymous, 1998. 1998 Guidelines for treatment of sexually transmitted diseases. Centers for disease control and prevention. MMWR Recomm. Rep., 47: 1-111.
PubMed - DuPont, H.L. and C.D. Ericsson, 1993. Prevention and treatment of traveler’s diarrhea. N. Engl. J. Med., 328: 1821-1827.
CrossRefPubMedDirect Link - Bennish, M.L., M.A. Salam, W.A. Khan and A.M. Khan, 1992. Treatment of shigellosis: III. Comparison of one- or two-dose ciprofloxacin with standard 5-day therapy. A randomized, blinded trial. Ann. Intern. Med., 117: 727-734.
CrossRefPubMedDirect Link - Khan, W.A., C. Seas, U. Dhar, M.A. Salam and M.L. Bennish, 1997. Treatment of shigellosis: V. Comparison of azithromycin and ciprofloxacin. A double-blind, randomized, controlled trial. Ann. Intern. Med., 126: 697-703.
CrossRefPubMedDirect Link - Miedouge, M., J. Hacini, F. Grimont and J. Watine, 2000. Shiga toxin-producing Escherichia coli urinary tract infection associated with hemolytic-uremic syndrome in an adult and possible adverse effect of ofloxacin therapy. Clin. Infect. Dis., 30: 395-396.
CrossRefPubMedDirect Link - Aubier, M., R. Verster, C. Regamey, P. Geslin and J.B. Vercken, 1998. Once-daily sparfloxacin versus high-dosage amoxicillin in the treatment of community-acquired, suspected pneumococcal pneumonia in adults. Sparfloxacin European Study Group. Clin. Infect. Dis., 26: 1312-1320.
CrossRefPubMedDirect Link - Chen, D.K., A. McGeer, J.C. de Azavedo and D.E. Low, 1999. Decreased susceptibility of Streptococcus pneumoniae to fluoroquinolones in Canada. N. Engl. J. Med., 341: 233-239.
CrossRefPubMedDirect Link - Wortmann, G.W. and S.P. Bennett, 1999. Fatal meningitis due to levofloxacin-resistant Streptococcus pneumoniae. Clin. Infect. Dis., 29: 1599-1600.
CrossRefPubMedDirect Link - Gentry, L.O. and G. Rodriguez-Gomez, 1991. Ofloxacin versus parenteral therapy for chronic osteomyelitis. Antimicrob. Agents Chemother., 35: 538-541.
CrossRefPubMedDirect Link - Chocarro, A., A. Gonzalez and I. Garcia, 2000. Treatment of tularemia with ciprofloxacin. Clin. Infect. Dis., 31: 623-624.
CrossRefPubMedDirect Link - Swartz, M.N., 2001. Recognition and management of anthrax-An update. N. Engl. J. Med., 345: 1621-1626.
CrossRefPubMedDirect Link - Schwartz, J., L. Jauregui, J. Lettieri and K. Bachmann, 1988. Impact of ciprofloxacin on theophylline clearance and steady-state concentrations in serum. Antimicrob. Agents Chemother., 32: 75-77.
CrossRefPubMedDirect Link - Halliwell, R.F., P.G. Davey and J.J. Lambert, 1993. Antagonism of GABAA receptors by 4-quinolones. J. Antimicrob. Chemother., 31: 457-462.
CrossRefPubMedDirect Link - Stein, G.E., 1996. Pharmacokinetics and pharmacodynamics of newer fluoroquinolones. Clin. Infect. Dis., 23: S19-S24.
CrossRefPubMedDirect Link - Burkhardt, J.E., J.N. Walterspiel and U.B. Schaad, 1997. Quinolone arthropathy in animals versus children. Clin. Infect. Dis., 25: 1196-1204.
CrossRefPubMedDirect Link - Cooper, C.S., P.L. Klock, D.T. Chu, D.J. Hardy, R.N. Swanson and J.J. Plattner, 1992. Preparation and in vitro and in vivo evaluation of quinolones with selective activity against gram-positive organisms. J. Med. Chem., 35: 1392-1398.
PubMed - Pan, X.S. and L.M. Fisher, 1998. DNA gyrase and topoisomerase IV are dual targets of clinafloxacin action in streptococcus pneumonia. Antimicrob. Agents Chemother., 42: 2810-2816.
PubMedDirect Link - Heddle, J. and A. Maxwell, 2002. Quinolone-binding pocket of DNA gyrase: Role of GyrB. Antimicrob. Agents Chemother., 46: 1805-1815.
CrossRefPubMedDirect Link - Boehm, H.J., M. Boehringer, D. Bur, H. Gmuender and W. Huber et al., 2000. Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods and 3D guided optimization. A promising alternative to random screening. J. Med. Chem., 43: 2664-2674.
CrossRefPubMedDirect Link - Schechner, M., F. Sirockin, R.H. Stote and A.P. Dejaegere, 2000. Functionality maps of the ATP-binding site of DNA gyrase-B: Generation of a consensus model of ligand binding. J. Med. Chem., 47: 4373-4390.
CrossRefPubMedDirect Link - Chatterji, M., S. Unniraman, S. Mahadevan and V. Nagaraja, 2001. Effect of different classes of inhibitors on DNA gyrase from Mycobacterium smegmatis. J. Antimicrob. Chemother., 48: 479-485.
CrossRefPubMedDirect Link - Jayashree, B.S., S. Thomas and Y. Nayak, 2010. Design and synthesis of 2-quinolones as antioxidants and antimicrobials: A rational approach. Med. Chem. Res., 19: 193-209.
CrossRefDirect Link - Joseph, B., F. Darro, A. Behard, B. Lesur and F. Collignon et al., 2002. 3-Aryl-2-quinolone derivatives: Synthesis and characterization of in vitro and in vivo antitumor effects with emphasis on a new therapeutical target connected with cell migration. J. Med. Chem., 45: 2543-2555.
CrossRefPubMedDirect Link