
D.K. Satnami
Natural Products Laboratory, Department of Chemistry, Dr. H.S. Gour University Sagar 470003 (M.P.) India
R.N. Yadava
Natural Products Laboratory, Department of Chemistry, Dr. H.S. Gour University Sagar 470003 (M.P.) India
Research Journal of Phytochemistry
Year: 2011 | Volume: 5 | Issue: 1 | Page No.: 22-31







ABSTRACT
This study described phytochemical examination of flowers of Caesalpinia crista Linn. Four flavonoid compounds have been characterized as 3, 5, 7, 3', 4'-pentahydroxy flavanone-3-O-β-D-xylopyranosyl-7-O-α-L-arabinopyranosyl-(1→3)-O-α-L-rhamnopyranoside (A), 4'-hydroxy-5, 7-dimethoxy flavone-4'-O-β-D-xylopyranosyl-(1→3)-O-β-D-glucopyranosyl-(1→4)-O-α-L-rhamnopy ranoside (B) 5, 2'-dihydroxy-6, 7-dimethoxy isoflavone (C) and 3, 5, 7, 3', 4' ,5'-hexahydroxy flavone (D) by various colour reactions, chemical degradations and spectral analysis from this plant. Acid hydrolysis, permethylation and enzymatic hydrolysis of compounds were carried out for identification of compounds. In this study were found to be isolated for the first time in this plant species. Isolated compounds were screened against S. enteritidis, K. pneumoniae, P. aeruginosa and M. lutens, bacteria and antifungal activity against F. oxisporum, A. niger and P. digitatum fungi showed good activity. Streptomycin used as standard antibacterial agent and Griseofulvin used as standard antifungal agent.
PDF Abstract XML References Citation
How to cite this article
D.K. Satnami and R.N. Yadava, 2011. Potential Phytochemical from Caesalpinia crista Linn. Research Journal of Phytochemistry, 5: 22-31.
URL: https://scialert.net/abstract/?doi=rjphyto.2011.22.31
URL: https://scialert.net/abstract/?doi=rjphyto.2011.22.31
INTRODUCTION
Plants have been and still are a rich source of many natural products in major part of India and other countries most of which have been extensively used for traditional human health care systems. The vast majority of people in the world takes care of themselves and uses healing plants that have been used for hundreds of generations. India is a country of vast biodiversity and traditional knowledge of using herbal medicines to cure many ailments (Cordell, 1995; Farnsworth and Soejarto, 1991; Taylor et al., 2001). Flavonoids are plant specific secondary metabolites, Compounds so named because they are not apparently involved in the survival of the cell. The basic flavonoid structure is the flavan nucleus which consists of 15 carbon atoms arranged in three rings (C6-C3-C6), which are labeled A, B and C from the degree structural variation emanates in part from the degree and pattern of hydroxylation. methoxzylation., prenylation, or glycosylation. These display a remarkable spectrum of biological activities including those that might be able to influence processes that are dysregulated during cancer development. these include, for example, antiallergic anti-inflammatory. antioxidant, antimutagenic antic-arcinogenic and modulation of enzymatic activities (Craig, 1999; Harborne and Williams, 2000). Caesalpinia crista Linn. (Asolkar et al., 2000) belongs to family Leguminosae (sub family Caesalpiniaceae). It is commonly known as Karanjava or karanju in Hindi. It is distributed throughout India and in most tropical countries. The root-bark is useful in treatment of tumour and for removing placenta. Its flower cures kapha and vata. The flowers are useful in treatment of colic, malaria, hydrocele, leprosy and skin diseases. The oil from the fruits is good for treatment of indolent ulcer. The seeds are very useful into treatment of antiperiodic, antipyretic and as tonic. The oil from the seeds is useful in convulsions and paralysis (Kirtikar and Basu, 1954). The plant family Leguminosae is taxonomically the second largest family of flowering plants comprising about 550 genera 1300 species of herbs, shrubs and tress. It’s divided into three sub families Papilionaceae, Caesalpiniaceae and Mimosaceae.
Earlier workers (Rastogi and Mehrotra, 2001; Peter and Tinto, 1997; Deon et al., 1998; Surya et al., 2004) have reported various constituents from this plant. In the present study, we report the isolation and structure elucidation of four compounds from methanolic extract of the flowers of this plant. Isolated compounds have been characterized as 3, 5, 7, 3', 4'-pentahydroxy flavanone-3-O-β-D-xylopyranosyl-7-O-α-L-arabinopyranosyl-(1→3)-O-α-L-rhamnopyranoside (A), 4'-hydroxy-5, 7-dimethoxy flavone-4'-O-β-D-xylopyranosyl-(1→3)-O-β-D-glucopyranosyl-(1→4)-O-α-L-rhamnopy ranoside (B) 5, 2'-dihydroxy-6, 7-dimethoxy isoflavone (C) and 3, 5, 7, 3', 4', 5'-hexahydroxy flavone. This study describes the phytochemical investigation of Caesalpinia crista Linn and spectroscopic techniques were used in elucidation of structure of compounds.
MATERIALS AND METHODS
General experimental procedure: All the melting points were determined on a thermoelectrically melting point apparatus and are uncorrected. The IR Spectra were recorded in KBr disc. UV Spectra was determined on Shimadzu-120 double beam spectrophotometer in MeOH. The IR Spectra were recorded on Shimadzu FTIR-8400 spectrophotometer in KBr disc. 1H-NMR Spectra were recorded on Varian XL 300 MHz spectrometer in CDCl3 using TMS as internal standard. 13C-NMR Spectra were recorded on Varian XL 90 MHz spectrometer using CDCl3. The chemical shift values are reported in ppm (δ) units and coupling constant (J) in Hz. The FAB mass spectra were recorded on a JEOL SX -102/DA-6000 Mass Spectrometer /Data System using Argon/Xenon (6 kv) as the FAB gas. Paper chromatography on Whatman charomatography paper No.1, thickness 0.16 mm were used. TLC on silica gel 60 F254 (0.5 mm thickness, glass plate, Merck) and column chromatography on silica gel (silica gel 60 mesh 0.040-0.063 nm, Merck) were used.
Plant materials: The flowers of Caesalpinia crista Linn. were procured from Sagar region into July-2008 and were taxonomically authenticated by the Department of Botany, Dr. H.S. Gour University Sagar. A voucher specimen has been deposited (No. DK/30/15 July 2008) in the Natural Products Laboratory, Department of Chemistry, Dr. H.S. Gour University, Sagar (M.P.) INDIA.
Extraction and isolation: Air dried and powdered seeds (5.6 kg) of the plant were extracted with petroleum ether (40-60°C) in Soxhlet extractor for 4 days. The flowers were successively extracted with methanol for three days. The MeOH soluble fraction of the plant was concentrated under reduced pressure, to yield a light brown viscous mass (3.5 g) which was subjected to TLC examination over silica gel-G using nBAW (4:1:5) as solvent and I2 vapors as visualizing agent, showed four spots, indicating it to be a mixture of four compounds A, B, C and D. These compounds were separated and purified by column chromatography over silica gel using CHCl3: MeOH in various proportions (0:10, 2:8, 6:4 and 4:10). After removal of the solvent and crystallization from ether, above eluates yielded compound A (0.71 g), compound B (0.58 g), compound C (0.45 g) and compound D (0.38 g), respectively.
RESULTS AND DISCUSSION
Phytochemical investigation of flowers of Caesalpinia crista Linn led to isolation of four flavonoids 3, 5, 7, 3', 4'-pentahydroxy flavanone-3-O-β-D-xylopyranosyl-7-O-α-L-arabinopyra nosyl-(1→3)-O-α-L-rhamnopyranoside (A) 4'-hydroxy-5, 7-dimethoxyflavone-4'-O-β-D-xylopy ranosyl-(1→3)-O-β-D-glucopyranosyl-(1→4)-O-α-L-rhamnopyranoside (B), 5, 2'-dihydro xy-6, 7-dimethoxy isoflavone (C) and 3, 5, 7, 3', 4', 5'-hexahydroxy flavone (D) from the methanol soluble fraction using coloum chromatography.
The structures of the isolated compound were stabilized by elemental anlysis UV, IR, 1H-NMR, 13C-NMR spectroscopy and mass spectrometry. Complete acid hydrolysis, permethylation and enzymatic hydrolysis of flavonoid glycosides A and B was carried out to yielded aglycone and sugar moieties, respectively.
Study of compound A: It has yellow amorphous power, m. p. 245-246°C m. f. C31H38O19 [M+] 714 (FABMS), Found (%) C 52.15, H 5.36, Calcd for m. f. C31H38O19, C 52.10, H 5.32. UV (MeOH) λmax nm; 278, 365. IR (KBr) vmax (cm-1); 3420, 2905, 1735, 1655, 1650, 1516, 1475, 1275, 1068, 830. 1HNMR (300 MHz, CDCl3) δ ); 11.72 (1H, s, 5-OH), 9.85 (1H, s, 3'-OH), 9.98 (1H, s, 4'-OH), 5.56 (1H, d, J Hz 10.2 H-2), 4.95 (1H, d, J Hz 10.5 H-3), 5.74 (1H, d, J 2.2 Hz, H-6), 5.99 (1H, d, J 2.2 Hz, H-8), 6.92 (1H, d, J 2.3 Hz, H-2'), 6.85 (1H, d, J 8.1 Hz, H-5'), 6.74 (1H, d, J 8.2, Hz, H-6'), 5.43 (1H, d, J 7.0 Hz H-1''), 4.32 (1H, d, J 2.1 Hz,, H-2''), 3.49 (1H, m, H-3''), 3.52 (1H, m, H-4''), 3.78 (1H, m, H-5''), 5.57 (1H, d, J 1.6 Hz, H-1'''), 4.03 (1H, dd, J 1.6, 3.6, Hz, H-2'''), 3.82 (1H, dd, J 3.6, 9.0 Hz, H-3'''), 3.48 (1H, d, J 9.0 Hz, H-4'''), 3.58 (1H, m, 5'''), 1.26 (3H, d, J 5.6 Hz, H-6'''), 5.52 (1H, d, J 1.9 Hz, H-1''''), 3.54 (1H, m, H-2''''), 3.44 (1H, m, H-3''''), 3.72 (1H, m, H-4''''), 3.78 (1H, m, H-5'''', 4.18 (1H, m, H-6''''). 13CNMR (90 MHz, CDCl3); δ 89.1 (C-2), 77.6 (C-3), 198.4 (C-4), 165.3 (C-5), 97.9 (C-6), 168.4 (C-7), 98.9 (C-8), 163.7 (C-9), 102.5 (C-10), 128.9 (C-1'), 116. 5 (C-2'), 147.1 (C-3'), 147.0 (C-4'), 115.8 (C-5'), 121.0 (C-6'), 108.0 (C-1''), 81.2 (C-2''), 76.9 (C-3''), 88.6 (C-4''), 65.2 (C-5''), 104.0 (C-1'''), 78.2 (C-3'''), 77.9 (C-5'''), 75.6 (C-2'''), 71.2 (C-4'''), 62.5 (C-6'''), 101.0 (C-1''''), 71.2 (C-2''''), 72.9 (C-3''''), 68.6 (C-4''''), 73.2 (C-5'''').
714 [M]+ , 582 [M+ - xylose], 450 [M+- xylose-arabinose ], 304 [xylose-arabinose-rhamnose] + (aglycone).
Acid hydrolysis of compound A: Compound A (75 mg) was dissolved in ethanol (15 mL) and refluxed with 10% H2SO4 (10 mL) on water bath for 7 h. The contents were concentrated and allowed to cool and residue was extracted with diethyl ether. The ethereal layer was washed with water and the residue was chromatographed over silica gel using CHCl3: MeOH as solvent to give aglycone A-1, which was identified as 3, 5, 7, 3', 4'-pentahydroxy flavanone by comparison of its spectral data with reported literature values14. The aqueous hydrolysate was neutralized with BaCO3 and the BaSO4 was filtered off. The filtrate was concentrated and subjected to paper chromatography examination using nBAW (4:1:5) solvent system and sugar were identified as D-xylose (Rf 0.25) L-arabinose (Rf 0.22) and L-rhamnose (Rf 0.36) (by Co-PC and Co-TLC).
Permethylation of compound A: Compound A (35 mg) was dissolved in DMF (50 mL) and treated with MeI (5 mL) and Ag2O (25 mg) in a 150 mL round bottomed flask fitted with condenser and refluxed for 2 days. The contents were filtered and washed with DMF. The filtrate was concentrated under reduced pressure and hydrolysed with 10% H2SO4 to give methylated aglycone, identified as 3, 7-dihydroxy- 5, 3', 4', -trimethoxy flavanone. The aqueous hydrolysate obtained after the removal of aglycone was neutralized with BaCO3 and the BaSO4 was filtered off. The filtrate was concentrated under reduced pressure and subjected to paper chromatography on Whatmann filter paper No.1 using n-BAW (4:1:5) as solvent system and aniline hydrogen phthalate as spraying agent. The methylated sugars were identified as 2, 3, 4-tri-O-methyl-D-xylose, 2, 3, 4-tri-O-methyl-L-arabinose and 2, 4, -di-O-methyl-L-rhamnose (by m.m.p and co-pc).
Enzymatic hydrolysis of compound A: Compound A (40 mg) was dissolved in MeOH (25 mL) and hydrolyzed with equal volume of almond emulsin enzyme. The reaction mixture was allowed to stay at room temperature for two days and filtered. The hydrolysate was concentrated and subjected to paper chromatography examination using nBAW (4:1:5) as solvent system and aniline hydrogen phthalate as a spraying reagent which showed the presence of D-xylose (R f 0.26). The proaglycone was dissolved in MeOH (20 mL) further hydrolyzed with equal volume of takadiastase at room temperature as usual procedure yielded aglycone identified as 3, 5, 7, 3', 4'-pentahydroxy flavanone and sugars were identified as L-arabinose (Rf 0.22) and L-rhamnose (Rf 0.36) (Co-PC and Co-TLC).
Study of compound A-1: It has yellow power, m.p.224-225°C m. f. C15H12O7 [M+] 304 Found (%) C 59.19, H 3.90, Calcd for m. f. C15H12O7, C 59.21, H 3.94. UV (MeOH) λmax nm; 285, 330. IR (KBr) vmax (cm-1); 3425, 1655, 2925, 1705, 1610, 1516, 1475, 1275, 1075, 835. 1HNMR (300 MHz, CDCl3): δ ); δ 11.90 (1H, s, 3-OH),11.76 (1H, s, 5-OH), 10.52 (1H, s, 7-OH), 9.78 (1H, s, 3'-OH), 9.33 (1H, s, 4'-OH), 5.62 (1H, d, J 10.5 Hz, H-2), 4.88 (1H, d, J 10.9, Hz, H-3),5.71 (1H, d, J 2.2 Hz, H-6), 5.94 (1H, d, J 2.2 Hz, H-8), 6.90 (1H, d, J 2.3 Hz, H-2'), 6.72 (1H, d, J 8.1 Hz, H-5'), 6.80 (1H, d, J 8.2, Hz, H-6'). 13CNMR (90 MHz, CDCl3 ); δ 85.2 (C-2), 77.0 (C-3), 199.0 (C-4), 165.5 (C-5), 97.6 (C-6), 168.0 (C-7), 98.4 (C-8), 164.2 (C-9), 101.9 (C-10), 128.7 (C-1'), 115.4 (C-2'), 145.3 (C-3'), 147.5 (C-4'), 115.6 (C-5'), 120.1 (C-6').
Study of compound B: It has yellow amorphous power, m. p.220-222OC, m. f. C34H42O18, [M+] 738, Found (%) C 55.25, H 5.66, Calcd for m. f. C34H42O18, C 55.28, H 5.69. UV (MeOH) λmax nm; 268, 375. IR (KBr) vmax (cm-1); 3392, 2978, 2846, 2683, 1745, 1665, 1609, 1495, 1452, 1272 1025, 814. 1HNMR (300 MHz, CDCl3); δ 3.44 (5-OMe), 3.56 (7-OMe), 10.09 (1H, s, 4'-OH), 6.58 (1H, s, H-3), 6.52 (1H, d, J 11.2Hz, H-6 ), 6.78 (1H, d, J 1.2 Hz, H-8 ), 7.01 (1H, d, J 9.1 Hz, H-2'), 6.94 (1H, d, J 9.1 Hz, H-6'), 6.91 (1H, d, J 9.2 Hz,, H-3'), 6.97 (1H, d, J 9.4 Hz,, H-5'), 5.56 (1H, d, J 2.01 Hz, H-1''), 4.03 (1H, dd, J 1.6, 3.6, Hz, H-2''), 3.82 (1H, dd, J 3.6, 9.0 Hz, H-3''), 3.48 (1H, d, J 9.0 Hz, H-4''), 3.58 (1H, m, 5''), 1.26 (3H, d, J 5.6 Hz, H-6''), 5.47 (1H, d, J 7.6 Hz, H-1'''), 3.54 (1H, m, H-2'''), 3.44 (1H, m, H-3'''), 3.72 (1H, m, H-4'''), 3.78 (1H, m, H-5'''), 4.18 (1H, m, H-6'''), 4.76 (1H, d, J 7.2 Hz, H-1''''), 3.24 (1H, m, H-2''''), 3.30 (1H, m, H-3''''), 3.32 (1H, m, H-4''''), 3.28 (1H, m, H-5''''). 13C-NMR (90 MHz, CDCl3 ); δ 163.0 (C-2), 104.7 (C-3), 176.2 (C-4), 162.8 (C-5), 98.1 (C-6), 164.5 (C-7), 97.8 (C-8), 160.0 (C-9), 105.1 (C-10), 122.7 (C-1'), 127.3 (C-2'), 115.6 (C-3'), 117.3 (C-4'), 115.9 (C-5'), 127.6 (C-6'), 104.7 (C-1''), 78.5 (C-3''), 78.2 (C-5''), 75.4 (C-2''), 70.8 (C-4''), 63.9 (C-6''), 101.2 (C-1'''), 71.6 (C-2'''), 72.9 (C-3'''), 68.3 (C-4'''), 73.4 (C-5'''), 63.1 (C-6'''), 103.5 (C-1''''), 76.7 (C-2''''), 73.4 (C-3''''), 69.9 (C-4''''), 66.4 (C-5''''), 56.2 (5-OMe), 54.9 (7-OMe).738 [M] +, 606 [M+-xylose], 444 [M+-xylose-glucose], 298 [M+-xylose-glucose-rhamnose]+ (aglycone).
Acid hydrolysis, permethylation and enzymatic hydrolysis of compound B was carried out by similar method as given for compound A.
Study of compound B-1: It has yellow power, m. p.288-289OC, m. f. C17H14O5, [M+] 298, Found (%) C 68.42, H 4.64, Calcd for m. f. C17H14O5, C 68.45, H 4.69. UV (MeOH) λmax nm; 266, 325. IR (KBr) vmax (cm-1); 3352, 2683, 1740, 1659, 1619, 1485, 1462, 1278 1035, 820. 1HNMR (300 MHz, CDCl3); δ 3.65 (5-OMe), 3.82 (7-OMe), 10.22 (1H, s, 4'-OH), 6.65 (1H, s, H-3 ), 6.45 (1H, d, J 11.2Hz, H-6 ), 6.85 (1H, d, J 1.2 Hz, H-8 ), 7.7 (1H, d, J 9.1 Hz,, H-2'), 7.8 (1H, d, J 9.1 Hz, H-6'), 6.88 (1H, d, J 9.2 Hz,, H-3'), 7.6 (1H, d, J 9.4 Hz,, H-5'). 13CNMR (90 MHz, CDCl3 ); δ 160.9 (C-2), 105.2 (C-3), 175.9 (C-4), 161.4 (C-5), 96.3 (C-6), 164.1 (C-7), 96.4 (C-8), 159.5 (C-9), 108.9 (C-10), 122.5 (C-1'), 126.6 (C-2'), 115.3 (C-3'), 117.0 (C-4'), 115.8 (C-5'), 126.5 (C-6'), 55.5 (5-OMe), 53.8 (7-OMe).
Study of compound C: It has yellow power, m. p.273-274OC, m. f. C17H14O6, [M+] 314 Found (%) C 64.91, H 4.43, Calcd for m. f. C17H14O6, C 64.95, H 4.45. UV (MeOH) λmax nm; 272, 390 (+AlCl3); 310, 386. IR (KBr) vmax; 3450, 2975, 2838, 2276, 1735, 1644, 1563, 1325, 875, 820 cm-1. 1HNMR (300 MHz, CDCl3); δ 12.16 (1H, s, 5-OH), 9.34 (1H, s, 2'-OH), 3.85 (3H, s, 6-OMe), 3.92 (3H, s, 7-OMe), 7.1 (1H, d, J 7.5 Hz, H-3'), 7.4 (1H, dd, J 7.5,1.2 Hz, H-4'), 7.2 (1H, t, J 7.6 Hz, H-5'), 8.05 (1H, d, J 7.2, 1.5 Hz, H-6' ), 6.54 (1H, d, J 1.5 Hz, H-8). 13CNMR (90 MHz, CDCl3 ); δ 156.5 (C-2), 121.5 (C-3), 182.2 (C-4), 142.8 (C-5), 130.4 (C-6), 155.2 (C-7), 90.5 (C-8), 153.2 (C-9), 108.5 (C-10), 119.4 (C-1'), 157.4 (C-2'), 116.5 (C-3'), 130.4 (C-4'), 119.5 (C-5'), 132.5 (C-6'), 60.85 (6-OMe), 58.5 (7-OMe).
Study of compound D: It has yellow power, m. p. 229-230OC, m. f. C15H10O8, [M+] 318 Found (%) C 56.63, H 3.12, Calcd for m. f. C15H10O8 C 56.60, H 3.14. UV (MeOH) λmax nm; 265, 345 (+AlCl3) 315, 370 (+AlCl3/HCl) 313, 366. IR (KBr) vmax 3428, 2886, 2835, 1730, 1649, 1508, 1433, 1360, 1245, 1064, 850, 819 cm-1. 1HNMR (300 MHz, CDCl3); δ 11.23 (1H, s, 3-OH), 12.09 (1H, s, 5-OH), 11.89 (1H, s, 7-OH), 11.15 (1H, s, 3'-OH), 11.31 (1H, s, 4'-OH), 11.98 (1H, s, 5'-OH), 6.20 (1H, d, J 2.3 Hz, H-6 ), 6.54 (1H, d, J 2.2 Hz, H-8), 7.35 (1H, s, H-2' ), 7.31 (1H, s, H-6' ). 13CNMR (90 MHz, CDCl3 ); δ 147.5 (C-2), 135.8 (C-3), 170.2 (C-4), 104.5 (C-5), 158.6 (C-6), 100.1 (C-7), 166.2 (C-8), 96.5 (C-9), 163.1 (C-10), 125.4 (C-1'), 108.5 (C-2'), 147.0 (C-3'), 136.9 (C-4'), 145.8 (C-5'), 109.0 (C-6').
Compound A has m. p.245-246OC, m. f. C31H38O19, [M+] 714 (FABMS). It gave positive Molisch (Mann and Saunders, 1999; Shinoda 1928) tests showing its flavanoidal glycosidic nature. Its UV spectrum showed absorption bands at 278 and 365 nm suggesting it to be flavanone. Its IR spectrum showed bands at 3400 (-OH), 2905 (C-H saturated), 1738 (>C=O α-β unsaturated), 1610 (aromatic ring system), 1068 (glycosidic linkage), 835 cm-1. In 1H-NMR spectrum of compound A, five doublets 5.56 (1H, d, J 10.2 Hz), 4.95 (1H, d, J 10.5, Hz), δ 6.92 (1H, d, J 2.5 Hz), δ 6.85 (1H, d, J 8.0 Hz) and δ 6.74 (1H, d, J 8.2 Hz) were assigned to H-2, H-3 H-2', H-5’ and H-6’ respectively9. Two singlets at δ 5.74 and δ 5.99 due to H-6 and H-8 protons (Liang et al., 1993; Cordero et al., 2000). Three singlets at δ 11.72, δ 9.84 and δ 9.98 were assigned to -OH groups at C-5, C-3' and C-4' positions. The anomeric proton signals at δ 5.43 (1H, d, J 7.0 Hz, H-1''), δ 5.57 (1H, d, J 1.6 Hz, H-1''') and δ 5.52 (1H, d, J 1.9, Hz, H-1'''') were assigned to H-1'' of D-xylose and H-1'''of L-rhamnose and H-1'''' of L-arabinose respectively. In 1H-NMR spectrum, coupling constant of J1,2 (7.0 Hz) value of anomeric proton of D-xylose, confirmed β configuration of D-xylose while coupling constants of J1,2 (1.6 Hz) and J1,2 (1.9 Hz) values of anomeric protons of L-rhamnose and L-arabinse confirmed α configuration of L-rhamnose and L-arabinse (Rao et al., 1999; He et al., 2005).
Acid hydrolysis of compound A with 10% ethanolic H2SO4 gave aglycone A-1, m. p. 224-225°C, m.f. C15H12O7, [M]+ 304 (FABMS), identified as 3, 5, 7, 3', 4'-pentahydroxy flavanone by comparison of its spectral data with reported literature values (Li et al., 2008; Mabry et al., 1970; Harborne and Mabry, 1982; Lederer and Lederer, 1947). The aqueous hydrolysate obtained was neutralized with BaCO3 and the BaSO4 was filtered off. The filtrate was concentrated and subjected to paper chromatography (Whatman charomatography paper No. 1, thickness 0.16 mm) showed the presence of D-xylose (Rf 0.25), L-arabinose (Rf 0.21) and L-rhamnose (Rf 0.37). Quantitative estimation of sugars carried out by the procedure of Mishra and Mohan Rao (1960), revealed that all the three sugars were present in equimolar ratio (1:1:1). Periodate oxidation of compound A, confirmed that all the sugars were present in the pyranose form (Hirst and Jones, 1949).
The position of the sugar moieties in compound A were determined by permethylation (Hokomorni) followed by acid hydrolysis, yielded methylated aglycone and methylated sugars. The methylated aglycone was identified as 3, 7-dihydroxy 5, 3', 4'-trimethoxy flavanone, confirmed that the C-3 -OH and C-7 -OH positions of aglycone were involved in glycosidation. The methylated sugars were identified as 2, 3, 4-tri-O-methyl-D-xylose 2, 3, 4-tri-O-methyl-L-arabinose and 2, 4-di-O-methyl-L-rhamnose by paper chromatography with authentic sample.
Therefore it was concluded that the C-1'''' -OH of L-arabinose was linked with C-3''' of L-rhamnose, C-1''' of L-rhamnose was attached with -OH group at C-7 position of aglycone and C-1'' of D-xylose was attached with -OH group at C-3 position of aglycone. The inter glycosidic linkage (1→3) was found between L-arbinose and L-rhmnose (Petek, 1965).
Enzymatic hydrolysis (Harborne, 1965) of compound A with almond emulsin liberated D-xylose indicating the presence of β-linkage between D-xylose and proaglycone. Proaglycone on further hydrolysis with takadiastase enzyme, liberated L-arabinose, followed by L-rhamnose, suggesting the presence of α -linkage between L-arabinose and L-rhamnose as well as between L-rhamnose and aglycone. Thus the compound A was identified as 3, 5, 7, 3', 4'-pentahydroxy flavanone-3-O-β-D-xylopyranosyl-7-O-α-L-arabinopyranosyl-(1→3)-O-α-L-rham nopyranoside.
Compound B has m. p.298-300OC, m. f. C34H42O18, [M+] 738 (FABMS). It gave positive Molisch and Shinoda tests showing its flavonoidal glycosidic nature. Its UV spectrum showed absorption bands at 268 and 375 nm suggesting its flavonoidal nature. Its IR spectrum showed absorption bands at 3392 (-OH), 2846 (-OMe) 2978 (C-H saturated), 1745 (>C=O α-β unsaturated), 1609 (aromatic ring system), 1028 (glycosidic linkage), 814 cm-1. In 1H-NMR spectrum of compound B, three singlets at δ 6.58, δ 6.52 and δ 6.78 were assigned to H-3, H-6 and H-8 respectively. four doublets at δ 7.01, δ 6.91, δ 6.97 and δ 6.94 were assigned to H-2', H-3', H-5' and H-6' respectively .Two signals at δ 3.44 and δ 3.56 were found due to two methoxy groups at C-5 and C-7 positions. The anomeric proton signals at δ 5.56 (1H, d, J 2.01 Hz, H-1''), δ 5.47 (1H, d, J 7.6 Hz, H-1''') and δ 4.76 (1H, d, J 7.2 Hz, H-1'''') were assigned to H-1'' of L-rhamnose, H-1''' of D-glucose and H-1'''' of D-xylose respectively. In 1H-NMR spectrum, coupling constants of J1,2 (7.2 Hz) and J1,2 (7.5 Hz) values of anomeric proton of D-xylose and D-glucose confirmed β configuration of D-xylose and D-glucose while coupling constants of J1,2 (2.01 Hz) value of anomeric proton of L-rhamnose confirmed the α configurations of L-rhamnose.
Acid hydrolysis of compound B with 10% ethanolic H2SO4 gave aglycone B-1, m. p. 287-288°C, m. f. C17H14O5, [M]+ 298 (FABMS), identified as 4'-hydroxy-5, 7-dimethoxy flavone by comparison of its spectral data with reported literature values (Kao et al., 2004). The aqueous hydrolysate was neutralized with BaCO3 and the BaSO4 was filtered off. The filtrate was concentrated and subjected to paper chromatography (Whatman charomatography paper No. 1, thickness 0.16 mm) showed the presence of D-xylose (Rf 0.25), D-glucose (Rf 0.18) and L-rhamnose (Rf 0.37). Quantitative estimation of sugars was carried out by the procedure of Mishra and Mohan Rao (1996), revealed that all the three sugars were present in equimolar ratio (1:1:1). Periodate oxidation of compound B, confirmed that all the sugars were present in the pyranose form.
The position of the sugar moieties in compound B were determined by permethylation followed by acid hydrolysis, yielded methylated aglycone and methylated sugars. The methylated aglycone was identified as 4'-hydroxy 5, 7-dimethoxy flavone confirmed that glycosidation was involved at C-4' -OH position of the aglycone. The methylated sugars were identified as 2, 3, 4-tri-O-methyl-D-xylose, 2, 4, 6-tri-O-methyl-D-glucose and 2, 3-di-O-methyl-L-rhamnose (by Co-PC). Therefore it was concluded that the C-1'''' -OH of D-xylose was linked with C-4''' of D-glucose, C-1''' -OH of D-glucose was attached with C-4'' position of L-rhamnose and C-1'' of L-rhamnose was attached with C-4' -OH position of aglycone. The interglycosidic linkages (1→3) and (1→4) were found between D-xylose and D-glucose as well as between D-glucose and L-rhamnose.
Enzymatic hydrolysis of compound B with almond emulsin liberated D-xylose followed by D-glucose suggesting the presence of β-linkage between D-xylose and D-glucose. Proaglycone was further hydrolysed with takadiastase liberated L-rhamnose suggesting the presence of α -linkage between L-rhamnose and aglycone. Therefore, the compound B was identified as 4'-hydroxy-5, 7-dimethoxyflavone-4'-O-β-D-xylopyranosyl-(1→3)-O-β-D-glucopyranosyl-(1→4)-O-α-L-rhamnopyranoside.
Compound C has m. f. C17H14O6, m. p. 273-274°C, [M]+ 314 (FABMS). It gave positive Shinoda tests showing its flavonoidal nature. Its UV spectrum showed absorption, at 272 and 392 nm suggesting it to be isoflavone. Its IR spectrum showed bands at 3350 (-OH), 2975 (C-H aromatic), 1735 (>C=O α-β unsaturated), 1605 (aromatic ring system), 875 and 820 cm-1. In 1H-NMR spectrum of compound C showed four proton singlets at δ 6.92 (1H, d, J 6.5 Hz), δ 6.95 (1H, d, J 6.8 Hz), δ 6.95 (1H, d, J 6.8 Hz) and δ 7.05 (1H, d, J 6.90 Hz) were assigned to H-3', H-4' and H-5', H-6', respectively. Two singlets at δ 3.84 and δ 3.92 confirmed the presence of -OMe groups at C-6 and C-7 positions. Thus, the compound C was characterized as 5, 2'-dihydroxy-6, 7-dimethoxy isoflavone by comparison of its spectral data with reported literature values (Ayatollahi et al., 2004).
Compound D has m. f. C15H10O8, m. p. 225-226oC, [M]+ 318 (FABMS). It gave positive Shinoda tests showing its flavonoidal nature. Its UV spectrum showed absorption, at 265 and 345 nm suggesting it to be flavone. Its IR spectrum showed bands at 3415 (-OH), 2854 (-Ome), 2885 (C-H aromatic), 1686 (>C=O α-β unsaturated), 1640 (aromatic ring system), 1064, 850 and 819 cm-1. In 1H-NMR spectrum of compound D showed two singlets at 6.23 (1H, d, J 2.0 Hz) and 6.51 (1H, d, J 2.2 Hz) were assigned H-6 and H-8 and singlets at δ 7.35 (1H, d, J 6.0 Hz) and δ 7.31 (1H, d, J 6.5 Hz) were assigned to H-2', H-6' protons respectively. Therefore compound C was characterized as 3, 5, 7, 3', 4', 5'-hexahydroxy flavone by comparison of its spectral data with reported literature values (David et al., 1996).
Antimicrobial activity of above compounds was evaluated against various bacteria and fungi. The results reported in Table 1 showed that Compound A was found highly active against S. enteritidis bacteria and less active against K. pneumoniae bacteria. Compound B was found highly active against K. pneumoniae bacteria and showed less activity against P. aeruginosa. Compound C more active against M. lutens bacteria less activity against S. enteritidis. Compound D showed good activity active against P. aeruginosa bacteria and less active against S. enteritidis. In case of antifungal activity, the compound A and B more active against Fusarium oxisporum.
| Table 1: | Antibacterial and antifungal activity of compounds |
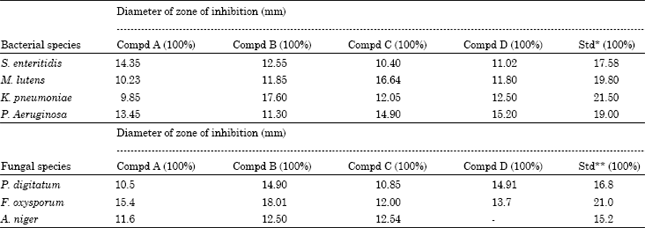 | |
| *Streptomycin used as standard antibacterial agent. **Griseofulvin used as standard antifungal agent | |
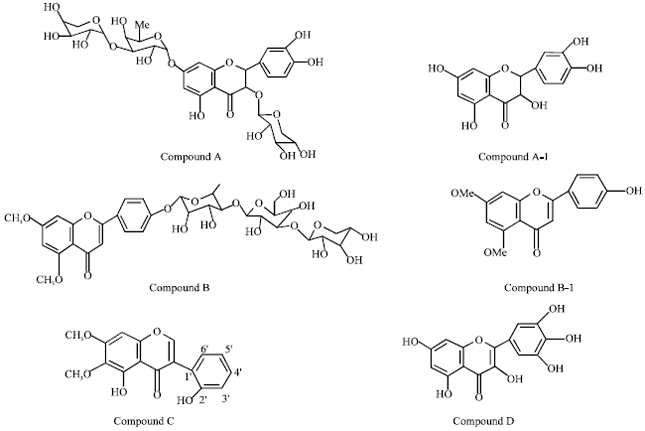 | |
| Fig. 1: | Flavonoid compounds isolated from Caesalpinia crista Linn. |
Compound C more active against Aspergillus’s niger. Compound D showed good activity against Penicellium digitatum and not active against Aspergillus’s niger fungi. Therefore these compounds may be used as therapeutic agent in diseases caused by these microorganisms (Fig. 1).
ANTIMICROBIAL ACTIVITY OF COMPOUNDS
The antimicrobial activity of compound A, B, C and D were determined by Filter Paper Disc Diffusion Method (Maruzella and Henry, 1958). The various bacterial species were first incubated at 45°C for 48 h. The sterile filter paper discs (6 mm) were soaked with standard antibacterial agent and various test samples and were dried at 50°C. The disc were then placed on soft nutrient agar (2%) petri plates previously seeded with suspension of each bacterial species. The diameters of zone of inhibition were measured at 37±1°C after 24 h. For fungal activity Saeboards broth media (Vincent and Vincent, 1944) with 4% agar was used for the preparation of plates and incubated with spores and mycelium suspension of fungi obtained from one week old culture. The diameter of zone of inhibition were measures at 28±1°C after 48 h. The results are recorded in Table 1.
ACKNOWLEDGMENTS
Authors are grateful to Head RSIC, Central Drug Research Institute, Lucknow (UP) India for recording various spectral data, elemental analysis and antimicrobial activity and also grateful to Head Department of Chemistry, Dr. H.S. Gour central University Sagar (M.P.) for providing necessary laboratory facilities. One of the author Dharmendra Kr. Satnami is thankful to UGC New Delhi for awarding Rajiv Gandhi National Research Fellowship (JRF 2006 and SRF 2008).
REFERENCES
- Cordell, G.A., 1995. Changing strategies in natural products chemistry. Phytochemistry, 40: 1585-1612.
CrossRef - Taylor, J.L.S., T. Rabe, L.J. McGaw, A.K. Jager and J. van Staden, 2001. Towards the scientific validation of traditional medicinal plants. Plant Growth Regul., 34: 23-37.
CrossRefDirect Link - Liang, H., L. Li and Y. Weimen, 1993. New flavonol glycoside from Epimedium acuminatum. J. Nat. Prod., 56: 943-945.
CrossRef - Cordero, C.M., M.L. Lazaro, J.L. Espartero and M.J. Ayuso, 2000. Retamatrioside, a new flavonol triglycoside from Retama sphaerocarpa. J. Nat. Prod., 63: 248-250.
CrossRef - Rao K.V., A.G. Damu, B. Jayaprakasham and D. Gunasekar, 1999. Flavonol glycosides from Cassia hirsuta. J. Nat. Prod., 62: 305-306.
PubMed - He, Z., C. Qiao, Q. Han, Y. Wang, W. Ye and H. Xu, 2005. New triterpenoid saponins from the roots of Platycodon grandiflorum. Tetrahedron, 61: 2211-2215.
CrossRef - Li, Y.L., J. Li, N.L. Wang and X.S. Yao, 2008. Flavonoids and new polyacetylene from Bidens parviflora Willd. Molecules, 13: 1931-1941.
PubMed - Mabry, T.J., K.R. Markham and M.B. Thomas, 1970. The Systematic Identification of Flavonoids. Springer Verlag, Berlin, Heidelberg, New York, Pages: 354.
Direct Link - Harborne, J.B., 1965. Plant polyphenols-XIV: Characterisation of flavonoid glycosides by acidic and enzymic hydrolysis. Phytochemistry, 4: 107-120.
CrossRef - Ayatollahi, S.M., M.R. Moein, F. Kobarfard and M.I. Choudhary, 2004. Two isoflavones from Iris songarica Scherenk. Daru, 12: 54-57.
Direct Link - Maruzella, J.C. and P.A. Henry, 1958. The antimicrobial activity of perfume oils. J. Am. Assoc. Sci., 47: 471-476.
CrossRef - Kalauni, S.K., S. Awale, Y. Tezuka, A.H. Banskota, T.Z. Linn and S. Kadota, 2004. Cassane and nor cassane type diterpenes of caesalpinia crista from Myanmar. J. Nat. Prod., 67: 1859-1863.
CrossRef - Harborne, J.B. and C.A. Williams, 2000. Advances in flavonoid research since 1992. Phytochemistry, 55: 481-504.
CrossRefPubMedDirect Link