
R. Javed
DNA Sequencing Lab, National Bureau of Animal Genetic Resources, Karnal-132001, Haryana, India
Mukesh
Department of Endangered Species Management, Wildlife Institute of India, Post Box 18, Chandrabani, Dehradun-248001, Uttarakhand, India
Pakistan Journal of Biological Sciences
Year: 2010 | Volume: 13 | Issue: 13 | Page No.: 657-663







ABSTRACT
Single Nucleotide Polymorphisms (SNPs) are the most frequent form of DNA variation in the genome. SNPs are genetic markers which are bi-allelic in nature and grow at a very fast rate. Current genomic databases contain information on several million SNPs. More than 6 million SNPs have been identified and the information is publicly available through the efforts of the SNP Consortium and others data bases. The NCBI plays a major role in facillating the identification and cataloging of SNPs through creation and maintenance of the public SNP database (dbSNP) by the biomedical community worldwide and stimulate many areas of biological research including the identification of the genetic components of disease. In this review article, we are compiling the existing SNP databases, research status and their application.
PDF Abstract XML References Citation
Received: March 06, 2010;
Accepted: May 23, 2010;
Published: July 08, 2010
How to cite this article
R. Javed and Mukesh, 2010. Current Research Status, Databases and Application of Single Nucleotide Polymorphism. Pakistan Journal of Biological Sciences, 13: 657-663.
DOI: 10.3923/pjbs.2010.657.663
URL: https://scialert.net/abstract/?doi=pjbs.2010.657.663
DOI: 10.3923/pjbs.2010.657.663
URL: https://scialert.net/abstract/?doi=pjbs.2010.657.663
INTRODUCTION
A single-nucleotide polymorphism (SNP, pronounced snip) is a DNA sequence variation occurring when a single nucleotide-A, T, C, or G- in the genome (or other shared sequence) differs between members of a species (or between paired chromosomes in an individual). For example, two sequenced DNA fragments from different individuals, AAGCCTA to AAGCTTA, contain a difference in a single nucleotide. In this case, there are two alleles-C and T. Almost all common SNPs have only two alleles. Single nucleotides may be changed (substitution), removed (deletions) or added (insertion) to a polynucleotide sequence. Ins/del SNP may shift translational frame. SNPs are mostly bi-allelic polymorphism and may occur in both coding and non coding region of the genome. The SNPs found with in a coding sequence are of particular interest of scientist because they are more likely to alter the biological function of a protein (Shen et al., 2006). A SNP has a minor allele frequency of greater than 1% at least one population (Risch, 2000). The SNPs are essential for personalized medicine (Chang et al., 2007; Suh and Vijg, 2005; Erichsen and Chanock, 2004). Many methodologies are reported or reviewed for genetic association studies (Lunn et al., 2006; Su et al., 2005). Database of SNPs provide a powerful resource for association studies that try to establish a relationship between phenotypes. However, SNPs may play a useful role in niche applications such as mitochondrial DNA (mt DNA), Y-SNPs, Ancestry Informative Markers (AIMs), predicting phenotypic traits and other potential forensic case work applications (Bulbul et al., 2009). Coding region SNPs can fulfill a useful role for separating common HV1/HV2 mitochondrial DNA types (Coble et al., 2004, 2006) and assays have been developed to reliably examine mt DNA coding region SNP variation (Brandstatter et al., 2003; Vallone and Butler, 2004; Vallone et al., 2004). A total number of 10-30 million SNPs in the human genome are estimated (Bostein and Risch, 2003).
Over the past few years, SNPs have been proposed as the next generation of markers for the identification of loci associated with complex diseases and for pharmacogenetic applications (Lander and Schork, 1994; Lander, 1996; Risch and Merikangas, 1996; Kruglyak, 1997; Schafer and Hawkins, 1998). Association studies can genotype a limited set of SNPs that contribute to common haplotypes, with haplotype tagging SNPs (ht.SNPs) (Stram et al., 2003). Several biotech companies started the race and took the initiative to isolate 60,000 or more SNPs to develop a whole genome SNP-based map (http://www.abbott.com/news/1997news/pr072897.htm).
The importance of SNPs in genetic studies comes from at least three different categories. First, SNPs can be used to reconstruct the history of genome. This is due to their abundance and most are inherited from one generation to the next, evolutionary stable, making them easier to follow in population studies. Studying the frequency and distribution of SNPs can lead to information on the evolution of the species. Second, SNPs can be directly responsible for genetic diseases since they may alter the genetic sequence of gene or of a regulatory region. Finally, SNPs may be utilized as markers to build the high-density genetic maps need to perform association studies (Orapan and Suthat, 2007), which locate and identify genes of functional importance. It has been proposed that a set of 3,000 bi-allelic SNP markers would be sufficient for whole-genome-mapping studies in humans; a map of 100,000 or more SNPs has been proposed as an ultimate goal to enable effective genetic mapping studies in large populations (Gibbs, 2003). The SNP databases are important resources for performing genetic linkage, association and admixture studies, both academic and commercial groups are developing large number of genome-wide SNP dataset. These dataset now contain over 12.6 million SNPs (Baye et al., 2009).
Status of current research: Current estimates are that SNPs occur as frequently as every 100-300 bases. This implies in an entire human genome there are approximately 10 to 30 million potential SNPs. More than 6 million SNPs have been identified and the information has been made publicly available through the efforts of the SNP consortium and others databases. Many of these SNPs have unknown associations. Compilation of public SNPs by NCBI (http://www.ncbi.nlm.nih.gov/projects/SNP/) has produced a subset of SNPs defined as a non-redundant set of markers that are used for annotation of reference genome sequence and are thus referred to as reference SNPs (rsSNPs). At present there are several SNP public databases (Dvornyk et al., 2004). The largest are dbSNP and HGVbase containing together several million SNPs (Aerts et al., 2002). There also exist other relatively small or specific SNP databases, e.g., TSC (Thorisson and Stein, 2003) JSNP (Hirakawa et al., 2002), HOWDY (Hirakawa 2002), GeneSNPs (Marsh et al., 2002). While the databases have been continuously expanded, the quality and completeness of the deposited SNP data remain to be of particular importance and to be assessed. New technologies make possible for the first time to genotype hundred of thousands of SNPs simultaneously. In present time data mining is a reasonable approach to investigating the number of SNPs that are informative for ancestry information (Baye et al., 2009). One can investigate distribution and density of SNPs across the genome by using HapMap, Affymetrix illumina SNP databases. The SNP databases in their current status might have some limitations for studies of complex disorders, especially in different ethnic groups, due to incomplete and/or uneven representation of SNPs and/or candidate genes in these groups (Dvornyk et al., 2004). Several new SNP genotyping techniques have been developed viz. Illumina Infinium Assay, Sequenom iPLEX, Illumina Golden Gate, APEX-2, Taqman assay. All these above genotyping techniques are primer extension based SNP genotyping techniques (Goelet et al., 1999; Syvanen, 2001). SNP-RFLPing assay is also a new web based SNP genotyping technique (Hsueh et al., 2006). Recently, SNP–Blast http://www.ncbi.nih.gov/SNP/snp_blastByOrg.cgi was developed by coupling the NCBI dbSNP (Sherry et al., 2001) with a BLAST program of NCBI. SNP-BLAST is designed to perform the BLAST function among various SNP databanks for many species.
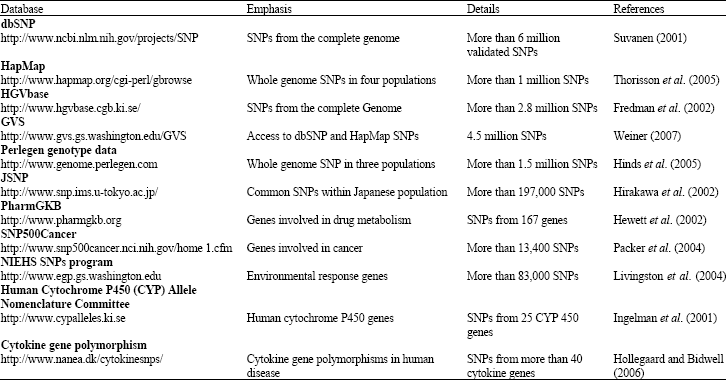
Available SNPs databases: In present days several SNP databases are available to search and choose the SNPs (Table 1).
The SNP consortium (TSC): The publicly funded efforts (NIH RFA: HG-98-001, 1998; (http://www.nhgri.nih.gov/Grant_info/Funding/rfa-hg-98-001.html) got a jump-start in 1999 when 13 pharmaceutical companies and the welcome trust formed the SNP Consortium (TSC) to accelerate SNP discovery and to ensure public accessibility to a minimum of 300,000 SNPs (http://www.snp.cshl.org/) (Lai, 2001). The goal was to generate a widely accepted, high-quality, extensive, publicly available map using SNPs as markers evenly distributed throughout the human genome. In the end, many more SNPs (1.8 million totals) were discovered (Thorisson and Stein, 2003). Now that the SNP discovery phase of the TSC project is essentially complete, emphasis has shifted to studying SNPs in populations. Various TSC member laboratories are genotyping a subset of SNPs as part of the Allele frequency project. The goal of the TSC allele frequency/genotype project is to determine the frequency of certain SNPs in three major world populations (http://www.snp.cshl.org). SNP data were made available through the consortium website at quarterly intervals during the project’s first year and at monthly intervals during the second year. This cycle of releases ceased in fall 2001 once the discovery phase was finished, but with recent additions of genotype and allele frequency information, new data were released in fall 2002.
Members of the SNP consortium: The international member companies, which together committed at least $30 million to the consortium’s efforts, are AP Biotech, AstraZeneca Group PLC, Aventis, Bayer Group AG, Bristol-Myers Squibb Co., F. Hoffmann-La Roche, Glaxo Wellcome PLC, IBM, Motorola, Novartis AG, Pfizer Inc., Searle and SmithKline Beecham PLC.
| Table 1: | Presently available SNP databases for Human genome and SNPs involved in diseases |
 | |
The Wellcome Trust contributed at least $14 million. Laboratories funded by these companies to identify SNPs are located at the Whitehead Institute, Sanger Centre, Washington University (St. Louis) and Stanford University. Data management and analysis take place at Cold Spring Harbor Laboratory.
The Single Nucleotide Polymorphism database (dbSNP): The National Center for Biotechnology Information (NCBI) has established the public SNP database dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and plays a major role in facilitating the identification and cataloging of SNPs through its creation and maintenance of the dbSNP database (Sherry et al., 2001). Database submissions can include a broad range of molecular polymorphisms: single base nucleotide substitutions, short deletion and insertion polymorphisms, microsatellite markers and polymorphic insertion elements such as retrotransposons (Benson et al., 1999). Once such variations are identified and catalogued in the database, additional laboratories can use the sequence information around the polymorphism and the specific experimental conditions for further research applications (Smigielski et al., 2000).
To facilitate research efforts, NCBI’s dbSNP is included in the Entrez retrieval system which provides integrated access to a number of software tools and databases that can aid in SNP analysis. For example, each SNP record in the database links to additional resources within NCBI’s Discovery Space (Wei et al., 2003) as shown in Fig. 1.
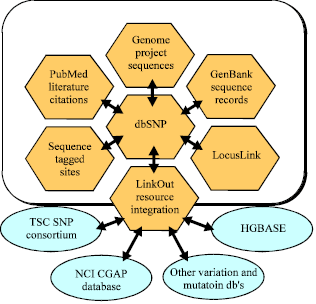 | |
| Fig. 1: | NCBI’s discovery space facilitating SNP research |
Resources include: GenBank, NIH’s sequence database; Entrez Gene, a focal point for genes and associated information; dbSTS, NCBI’s resource containing sequence and mapping data on short genomic landmarks; human genome sequencing data and PubMed, NCBI’s literature search and retrieval system. SNP records also link to various external allied resources. SNPs are also thought to be key enablers in realizing the concept of personalized medicine (The United States Congress defines personalized medicine as the application of genomic and molecular data to better target the delivery of health care, facilitate the discovery and clinical testing of new products). However, their greatest importance in biomedical research is for comparing regions of the genome between cohorts (In statistics and demography, a cohort is a group of subjects who have shared a particular experience during a particular time span).
SNPedia: SNPedia (pronounced snipedia) is a wiki-based bioinformatics web site that serves as a database of SNPs (Atanassova, 2009). Each article on a SNP provides a short description, links to scientific articles and personal genomics web sites, as well as microarray information about that SNP (Cariaso, 2007) http://www.bio-itworld.com/issues/2007/dec-jan/inside-the-box-snpedia/. Thus SNPedia may support the interpretation of results of personal genotyping from, e.g., 23 and Me, Navigenics, deCODEme or Knome.
SNPs applications
SNPs and disease diagnosis: The SNPs have been used as molecular markers in many studies of monogenic or complex diseases owing to their high frequency and simple (binary) variation pattern (Kim and Mishra, 2007). Each person’s genetic material contains a unique SNP pattern that is made up of many different genetic variations (Mircea et al., 2005). Kang et al. (2007) screened and scanned mitochondrial gene polymorphisms to determine the significance of mitochondrial DNA mutations in Korean migraineurs. According to Orapan and Suthat (2007) occasionally, a SNP may actually cause a disease and therefore, can be used to search for and isolate the disease-causing gene.
SNPs in association studies: Comparison of disease affected and unaffected group of individuals DNA for SNP patterns is called associate on study (Howard, 2005). Through association studies we can detect differences between the SNP patterns of the two groups; thereby indicating which pattern is most likely associated with the disease-causing gene (Mircea et al., 2005).
An example is the gene associated with the late onset Alzheimer’s apolipoprotein E or ApoE (Strittmatter and Roses, 1996). This gene contains two SNP that results in three possible alleles for the gene E2, E3 and E4. Most SNPs are not responsible for a disease state insteated; they serve as biological markers for pinpointing diseases. Because they are located near a gene found to be associated with certain disease (Barnes and Gray, 2003; Chakravarti, 2001). An association study of a major autism linkage region on human chromosome 17 was done by Stone et al. (2007). According to Matthias (2004), only 10% of all gene-based SNPs have sequence-predicted functional relevance making them a primary target for genotyping in association studies.
SNPs and drug development: Single Nucleotide Polymorphism (SNP) technologies can be used to identify disease-causing genes in humans and to understand the inter-individual variation in drug response. By establishing an association between the genetic make-up of an individual and drug response it may be possible to develop a genome-based diet and medicines that are more effective and safer for each individual (Shastry, 2007). In a recent study, Tang et al. (2005) analyzed genetic data from 3,636 people who identified themselves as belonging to one of four major racial and ethnic groups in the United States (Caucasian, African American, East Asian and Hispanic). The appeal of pharmacogenetics lies in the possibility of personalized medicine (Hunt, 2008).
SNPs in paternity investigation: Based on the 52 SNP-plex developed by the SNPforID Consortium, Costa et al. (2008) designed two 10-plex to study Single Nucleotide Polymorphisms (SNPs) for human identification and to establish its usefulness in paternity casework. Twenty autosomal SNP set was studied in 56 paternity investigation cases from south Portuguese resident population, also analyzed with 17 Short Tandem Repeats (STRs). The use of SNPs as a complement to the analysis of autosomal STRs in paternity casework can result in paternity index and paternity probability values equivalent or higher than those obtained with more STR loci, but with lower costs. Instead of using additional STR loci, the analysis of 20 autosomal SNPs, as a complement technique to standard methodologies, is an appealing alternative in paternity investigation cases (Paulo et al., 2009). Forty autosomal most informative and population-independent SNPs were used by Whittle et al. (2009) in paternity investigation and as a prelude for forensic human identification.
SNPs in functional proteomics and gene therapy: SNP related functional proteomics involve the identification of functional SNPs that modify proteins and protein active sites structure and function. Several studies have attempted to predict functional impact by mapping nsSNPs onto protein structures and to discover properties of structural context that distinguish disease-associated and neutral nsSNPs (Stitziel et al., 2003; Sunyaev et al., 2001; Wang and Moult, 2001). Functional proteomics is closely tied to modern (post-genomic) drug design and function SNP information helps to discover new therapeutic targets. Most interestingly, by developing a database of the modifications generated by functional (coding) SNPs in disease related proteins, new compounds can be designed for correcting or enhancing the effects of those mutations in the population.
SNPs in forensic field: Presently SNPs markers are playing a major role in forensic investigation. In Europe for the study of west European Caucasian haplogroups, 16 SNPs markers were selected from the coding region of the human mitochondrial DNA (Brandstatter et al., 2003).
CONCLUSIONS
Single Nucleotide Polymorphisms (SNPs) are the most frequent form of DNA variation in the genome. At present there are several SNP public databases in which dbSNP and HGVbase containing together several million SNPs. There also exist other relatively small or specific SNP databases, e.g., TSC, JSNP, HOWDY GeneSNPs. New technologies make possible for the first time to genotype hundred of thousands of SNPs simultaneously. Several new SNP genotyping techniques have been developed viz. Illumina Infinium Assay, Sequenom iPLEX, Illumina Golden Gate, APEX-2, Taqman assay. Many of these technologies have been efficiently used in large-scale association studies. However, even with this remarkable advancement, further improvements in existing technologies, as well as development of new methods, are still necessary for routine genome-wideSNP analyses in large sample collections, which must be cost-effective and time-efficient. SNP-RFLPing assay is also a new web based SNP genotyping technique. Recently, SNP-Blast was developed by coupling the NCBI dbSNP with a BLAST program of NCBI. SNP-BLAST is designed to perform the BLAST function among various SNP databanks for many species. SNPedia is also a wiki-based bioinformatics web site that serves as a database of SNPs. Presently SNP marker is very useful for biomedical fields like drug development, paternity investigation and gene therapy and most efficiently using in association studies.
Summary of point
| • | SNPs can search from the available databases for further studies, in which NCBI dbSNP is a major database |
| • | Several new SNP genotyping technologies are developed for genotyping of huge number of SNPs |
| • | Currently SNPs are very authoritative for several biomedical fields and most efficient for association studies |
ACKNOWLEDGMENTS
The authors are hearty thankful to Dr. M.S. Tantia for critical reading and valuable comments and Dr. R.K. Vijh for helpful suggestions and kind support to write this study. Authors also acknowledge the moral boost up provided by our colleagues.
REFERENCES
- Aerts, J., Y. Wetzels, N. Cohen and J. Aerssens, 2002. Data mining of public SNP databases for the selection of intragenic SNPs. Hum. Mutatat, 20: 162-173.
PubMed - Shastry, B.S., 2007. SNPs in disease gene mapping, medicinal drug development and evolution. J. Hum. Genet., 52: 871-880.
CrossRef - Bostein, P.E and N. Risch, 2003. Discovering genotypes underling human phenotypes: Past successes for Mendelian disease, future approaches for complex disease. Nature Genet., 33: 228-237.
PubMed - Brandstatter, A., T.J. Parsons and W. Parson, 2003. Rapid screening of mtDNA coding region SNPs for the identification of west European Caucasian haplogroups. Int. J. Legal Med., 117: 291-298.
CrossRef - Bulbul, O., C. Philips, D. Argac, M.S. Shahzad and M. Fondevila et al., 2009. Internal validation of 29 autosomal SNPs Multiple using AB310 genetic Analyzer. Forensic Sci. Int. Genet. Suppl. Ser., 2: 129-130.
CrossRef - Coble, M.D., R.S. Just, J.E. O`Callaghan, I.H. Letmanyi, C.T. Peterson, J.A. Irwin and T.J. Parsons, 2004. Single nucleotide polymorphisms over the entire mtDNA genome that increase the power of forensic testing in Caucasians. Int. J. Legal Med., 118: 137-146.
PubMed - Coble, M.D., P.M. Vallone, R.S. Just, T.M. Diegoli, B.C. Smith and T.J. Parsons, 2006. Effective strategies for forensic analysis in the mitochondrial DNA coding region. Int. J. Legal Med., 120: 27-32.
PubMed - Costa, G., P. Dario, I. Lucasm, T. Ribeiro, R. Espinheira and H. Geada, 2008. Autosomal SNPs in paternity investigation. Forensic Sci. Int. Genet. Suppl. Ser., 1: 507-509.
CrossRef - Dvornyk, V., J.R. Long, D.H. Xiong, P.Y. Liu and LY. Zhao et al., 2004. Current limitation of SNP data from the public domain for studies of complex disorders: a test for ten candidate genes for obesity and osteoporosis. BMC Genomics, 5: 4-4.
Direct Link - Lai, E., 2001. Application of SNP technologies in medicine: lessons learned and future challenges. Genome Res., 11: 927-929.
CrossRefDirect Link - Erichsen, H.C. and S.J. Chanock, 2004. SNPs in cancer research and treatment. Br. J. Cancer, 90: 747-751.
CrossRef - Fredman, D., M. Siegfried, Y.P. Yuan, P. Bork, H. Lehvaslaiho and A.J. Brookes, 2002. HGVbase: A human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res., 30: 387-391.
PubMed - Thorisson, G.A. and L.D. Stein, 2003. The SNP consortium website: past, present and future. Nucleic Acids Res., 31: 124-127.
Direct Link - Hinds, D., L. Stuve, G. Nilsen, E. Halperin and E. Eskin, 2005. Whole genome patterns of common DNA variation in three human populations. Science, 307: 1072-1079.
CrossRef - Hirakawa, M., 2002. HOWDY: an integrated database system for human genome research. Nucleic Acids Res., 30: 152-157.
Direct Link - Hirakawa, M., T. Tanaka, Y. Hashimoto, M. Kuroda, T. Takagi and Y. Nakamura, 2002. JSNP: a database of common gene variations in the Japanese population. Nucleic Acids Res., 30: 158-162.
PubMed - Hollegaard, M.V. and J.L. Bidwell, 2006. Cytokine gene polymorphism in human disease: On-line databases, Supplement 3. Genes Immun., 7: 269-276.
Direct Link - Chang, H.W., Y.H. Cheng, T.C. Chen, C.S. Yang, L.Y. Chuang and C.H. Yang, 2007. A novel method providing exact SNP IDs from sequences. Int. J. Comput. Sci., 34: 17-22.
Direct Link - Ingelman, S.M., M. Oscarson, A. Daly, S. Garte and D. Nebert, 2001. Human cytochrome P-450 (CYP) genes: a web page for the nomenclature of alleles. Cancer Epidemiol. Biomarkers Prev., 10: 1307-1308.
Direct Link - Kang, L., S.T. Lee, I. Wooseok, S.C. Kim, K.S. Hun, B.K. Kim and M. Kim, 2007. Screening of the A11084G polymorphism and scanning of a mitochondrial genome SNP in korean migraineurs. J. Clin. Neurol., 3: 127-132.
Direct Link - Kruglyak, L., 1997. The use of a genetic map of biallelic markers in linkage studies. Nature Genet., 17: 21-24.
CrossRef - Lander, E.S. and N.J. Schork, 1994. Genetic dissection of complex traits. Science, 265: 2037-2048.
PubMed - Livingston, R., A. von Niederhausern,, A. Jegga, D. Crawford and C. Carlson et al., 2004. Pattern of sequence variation across 213 environmental response genes. Genome Res., 14: 1821-1831.
PubMed - Lunn, D.J., J.C. Whittaker and N. Best, 2006. A Bayesian toolkit for genetic association studies. Genet. Epidemiol., 30: 231-247.
PubMed - Marsh, S., P. Kwok and H.L. McLeod, 2002. SNP databases and pharmacogenetics: great start, but a long way to go. Hum. Mutat., 20: 174-179.
PubMed - Matthias, W., 2004. Target SNP selection in complex disease association studies. BMC Bioinformatics, 5: 92-92.
PubMed - Orapan, S. and F. Suthat, 2007. Genetic polymorphism and implications for human diseases. J. Med. Assoc. Thailand, 90: 394-398.
PubMed - Packer, B.R., M. Yeager, L. Burdett, R. Welch and M. Beerman et al., 2004. SNP 500 Cancer: A public resource for sequence validation and assay development for genetic variation in candidate genes. Nucleic Acids Res., 32: 528-532.
PubMed - Paulo, D., R. Teresa, E. Rosa and G. Helena, 2009. SNPs in paternity investigation: The simple future. Forensic Sci. Int. Genet. Suppl. Ser., 2: 127-128.
CrossRef - Risch, N. and K. Merikangas, 1996. The future of genetic studies of complex human diseases. Science, 273: 1516-1517.
CrossRefPubMedDirect Link - Risch, N.J., 2000. Searching for genetic determinants in the new millennium. Nature, 405: 847-856.
PubMed - Schafer, A.J. and J.R. Hawkins, 1998. DNA variation and the future of human genetics. Nat. Biotechnol., 16: 33-39.
CrossRef - Shen, J., P.L. Deinniger and H. Zhao, 2006. Application of computational algorithm tools to identify functional SNPs in cytokine genes. Cytokine, 35: 62-66.
PubMed - Sherry, S., M. Ward, M. Kholodov, J. Baker, L. Phan, E.M. Smigielski and K. Sirotkin, 2001. dbSNP: The NCBI database of genetic variation. Nucl. Acids Res., 29: 308-311.
PubMed - Smigielski, E.M., K. Sirotkin, M. Ward and S.T. Sherry, 2000. dbSNP: A database of single nucleotide polymorphisms. Nucl. Acids Res., 28: 352-355.
CrossRefDirect Link - Hunt, S.Y., 2008. Pharmacogenetics, personalized medicine and race. Nature Educ., 1: 6-6.
Direct Link - Stitziel, N.O., Y.Y. Tseng, D. Pervouchine, D. Goddeau, S. Kasif, J. Liang, 2003. Structural location of disease-associated SNPs. J. Mol. Biol., 327: 1021-1030.
PubMed - Stram, D.O., P.C. Leigh, P. Bretsky, M. Freedman and J.N. Hirschhorn et al., 2003. Modeling and E-M estimation of haplotype- specific relative risks from genotype data for a case-control study of unrelated individuals. Hum. Heredity, 55: 179-190.
PubMed - Strittmatter, W.J. and A.D. Roses, 1996. Apolipoprotein E and alzheimer`s disease. Annual Rev. Neurosci., 19: 53-77.
PubMed - Su, S.C., C.C. Kuo and T. Chen, 2005. Inference of missingSNPs and information quantity measurements for haplotype blocks. Bioinformatics, 21: 2001-2007.
PubMed - Suh, Y. and J. Vijg, 2005. SNP discovery in association genetic variation with human disease phenotypes. Mutat. J., 573: 41-53.
PubMed - Sunyaev, S., V. Ramensky, I. Koch, W. Lathe, A.S. Kondrashov and P. Bork, 2001. Prediction of deleterious human alleles. Hum. Mol. Genet., 10: 591-597.
Direct Link - Syvanen, A.C., 2001. Accessing genetic variation: Genotyping single nucleotide polymorphisms. Nat. Rev. Genet., 2: 930-942.
CrossRef - Tang, H., T. Quertermous, B. Rodriguez, S.L.R. Kardia and X. Zhu, 2005. Genetic structure, self-identified race/ethnicity and confounding in case-control association studies. Am. J. Hum. Genet., 76: 268-275.
Direct Link - Baye, T.M., K.T. Tiwari, D.B. Allison, R.C. Go, 2009. Database mining for selection of SNP markers useful in admixture mapping. BioData Mining, 2: 1-1.
Direct Link - Thorisson, G., A. Smith, L. Krishnan and L. Stein, 2005. The international HapMap projectweb site. Genome Res., 15: 1592-1593.
CrossRef - Vallone, P.M. and J.M. Butler, 2004. Y-SNP typing of U.S. African American and Caucasian samples using allele-specific hybridization and primer extension. J. Forensic Sci., 49: 723-732.
Direct Link - Vallone, P.M., R.S. Just, M.D. Coble, J.M. Butler and T.J. Parsons, 2004. A multiplex allele-specific primer extension assay for forensically informative SNPs distributed throughout the mitochondrial genome. Int. J. Legal Med., 118: 147-157.
CrossRefPubMedDirect Link - Wang, Z. and J. Moult, 2001. SNPs, protein structure and disease. Hum. Mutat., 17: 263-270.
CrossRefPubMedDirect Link - Whittle, M.R., E.C. Favaro and D.R. Sumita, 2009. Paternity investigation experience with a 40 autosomal SNP panel. Forensic Sci. Int. Genet. Suppl. Ser., 2: 149-150.
CrossRef