
B.L. Bhaskara
Department of Chemistry, Amrita Vishwa Vidyapeetham, Bangalore Campus, Kasavanahalli, Carmelram Post, Bangalore 560035, India
S. Anil Kumar
Department of Chemistry, Amrita Vishwa Vidyapeetham, Bangalore Campus, Kasavanahalli, Carmelram Post, Bangalore 560035, India
U.R. Anil Kumar
Department of Chemistry, Manasagangotri, University of Mysore, India
Asian Journal of Applied Sciences
Year: 2011 | Volume: 4 | Issue: 3 | Page No.: 306-313







ABSTRACT
A new high performance liquid chromatography method is developed and validated to quantify atenolol in pharmaceutical preparations. The high performance liquid chromatography determination is performed on a reversed phase column, Atlantis dC18 (250x4.6 mm, 5 μm; internal diameter) using a mobile phase (1.0 mL min-1) with ultra violet detection at 225 nm. A rectilinear relationship between mean peak area and concentration of atenolol is observed in the range 1-100 μg mL-1 with a detection limit of 0.4 μg mL-1 and a quantitation limit of 1.0 μg mL-1. The validation parameters such as specificity, precision, linearity and range, ruggedness and robustness have been established according to the current ICH guidelines. The results were statistically compared with those of the reference/literature method by applying Student’s t-test and F-test. Accuracy, evaluated by means of the spike recovery method was in the range 98.3-102.5%. The method was characterized by a shorter retention time (3.39 min) and a wide linear dynamic range (1-100 μg mL-1) of concentration over which it is applicable. The method was demonstrated to be both accurate and precise which qualify it to be adopted for routine use in pharmaceutical quality control laboratories.
PDF Abstract XML References Citation
How to cite this article
B.L. Bhaskara, S. Anil Kumar and U.R. Anil Kumar, 2011. A Facile and Rapid HPLC Method for the Determination of Atenolol in Pharmaceutical Formulations. Asian Journal of Applied Sciences, 4: 306-313.
DOI: 10.3923/ajaps.2011.306.313
URL: https://scialert.net/abstract/?doi=ajaps.2011.306.313
DOI: 10.3923/ajaps.2011.306.313
URL: https://scialert.net/abstract/?doi=ajaps.2011.306.313
INTRODUCTION
Atenolol is a cardio selective β-adrenergic receptor-blocking agent without membrane-stabilizing or intrinsic sympathomimetic activities (Medical Economics, 1999). It is also used to treat myocardial infarction, arrhythmias, angina, disorders arising from decreased circulation and vascular constriction, including migraine (Wadworth et al., 1991). It is prescribed for high blood pressure and regulating tachycardia. It works by relaxing blood vessels and slowing heart rate to improve blood flow and decrease blood pressure (Fig. 1).
Symptoms of overdose may include unusually slow heartbeat, severe dizziness, shallow breathing, weakness or fainting. Atenolol is excreted in breast milk and may cause adverse effects in the breastfed infant (El-Gindy et al., 2008).
Many methods for the determination of atenolol have been reported such as Ultraviolet spectrophotometry (Ferraro et al., 2003; Wehner, 2000), high-performance liquid chromatography (Sasa et al., 1988; Ruiz-Angel et al., 2002), Capillary electrophoresis (Coragem Briguenti and Bonato, 2005; Shafaati and Clark, 1996), Ultra performance liquid chromatography (Wren and Tchelitcheff, 2006).
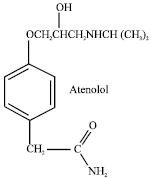 | |
| Fig. 1: | Structure of Atenolol - (RS)-4-(2-hydroxy-3- isopropylaminopropoxy) phenylacetamide |
Other known methods include spectrofluorimetry (Gajewska et al., 1992), kinetic methods using ammonium vanadate as oxidant (Sultan, 1992) differential scanning calorimetry and thermogravimetry (Phyramides et al., 1995), electrophoresis (Ferraro et al., 2004), NMR spectroscopy (Agarwal et al., 1992; Zakhari et al., 1991) and chemo- metric assisted spectrophotometric methods (El-Gindi et al., 2005).
Most of these reported methods are complicated and do require expensive instruments. Normal HPLC with UV or fluorescence detection need extraction steps and complicated clean up processes (Modamio et al., 1998; Zarapkar et al., 1997; Jain and Jain, 1991; Pawlak and Clark, 1992; Radulovic et al., 1991). The kinetic method is less sensitive and involves a heating step where as thermal methods require expensive experimental setup and also are less sensitive (Basavaiah et al., 2006).
This study deals with development and validation of a sensitive method for the assay of ATN in pharmaceuticals, based on HPLC technique. The separation and determination were caused on a reversed phase Atlantis dC18 column and UV-detection at 225 nm. The method was demonstrated to be both accurate and precise which qualify it to be adopted for routine use in pharmaceutical quality control laboratories.
MATERIALS AND METHODS
This study was conducted in 2010.
Apparatus: The chromatographic system consisted of an Agilent 1100 series chromatograph equipped with an in built solvent degasser, quaternary pump, photo diode array detector with variable injector and auto sampler and a reversed phase 5 μm Atlantis dC18 column (250x4.6 mm, internal diameter).
Reagents and standards: All chemicals used were of analytical reagent grade, Ammonium acetate (S.D. Fine Chem. Ltd, India) and HPLC grade methanol (Merck. Ltd, Mumbai) was used. Distilled water filtered through 0.45 μm filter (Millipore) was used to prepare solutions. Methanol and water in the ratio of 50:50 was used as a diluent for the sample preparations.
| • | Mobile phase A: 20 mM ammonium acetate |
| • | Mobile phase B: Methanol |
Pharmaceutical grade ATN, certified to be 99.85% pure was procured from Cipla India Ltd, Mumbai, India and was used as received. For the study, an accurately weighed 50 mg of ATN was dissolved in and diluted to volume with the diluent solution in a 100 mL calibrated flask to obtain a concentration of 500 μg mL-1 ATN.
Procedures
Chromatographic conditions: The separation was achieved at 30°C on the column using the mobile phase of 60:40 (20 mM ammonium acetate: methanol) at a flow rate of 1.0 mL min-1. The detector wavelength was set at 225 nm with a sensitivity of 0.2 a.u.f.s.
Calibration graph: Working standard solutions equivalent to 1 to 100 μg mL-1 of ATN were prepared by appropriate dilution of stock standard solution (500 μg mL-1) with the diluent solution. Ten μl aliquot of each solution was injected automatically on to the column in duplicate and the chromatograms were recorded. Calibration graph was prepared by plotting the mean peak area versus concentration of ATN.
The concentration of the unknown was read from the calibration graph or computed from the regression equation derived from the mean peak area-concentration data.
Assay in dosage forms: A quantity of tablet powder equivalent to 50 mg of ATN was accurately weighed into a 100 mL calibrated flask and 60 mL of diluent solution was added. The content was shaken for 20 min and then the volume was diluted to the mark and mixed well. A small portion of the extract (say 10 mL) was withdrawn and filtered through 0.2 μm filter to ensure the absence of particulate matter. The filtered solution was appropriately diluted with the diluent solution for analysis as described already.
RESULTS AND DISCUSSION
Method development: A solution of ATN was injected in duplicate on to the column and was monitored by UV-detection at 225 nm. An isocratic method was selected rather than gradient to get the lesser retention time. At a flow rate of 1.0 mL min-1, the retention time was 3.39 min (Fig. 2).
Under the described experimental conditions, the peak was well defined and free from tailing. ATN was determined by measuring the peak area. A plot of mean peak area against concentration gave a linear relationship (correlation coefficient, r = 0.9993, n = 5) over the concentration range 1-100 μg mL-1. Using the regression analysis, the linear equation, Y = (-2328) + (14857) X was obtained, where; Y is the mean peak area and X concentration in μg mL-1. The limits of detection and quantification calculated as per ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use., 2005) were 0.4 and 1.0 μg mL-1, respectively.
Method validation
Specificity: The specificity of the assay was examined by the parameters obtained in the chromatogram. The theoretical plates and tailing factor were found to be 5547 and 1.62, respectively. This indicates the system suitability of the method.
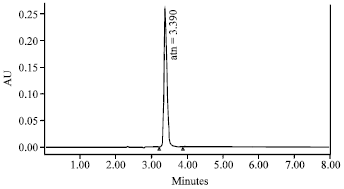 | |
| Fig. 2: | HPLC chromatogram of atenolol performed on a reversed phase column, Atlantis dC18 (250x4.6 mm, 5 μm; internal diameter) using 20 mM ammonium acetate and methanol as mobile phase (1.0 mL min-1) with ultra violet detection at 225 nm |
| Table 1: | Accuracy and intra-day precision |
 | |
| RE: Relative error and RSD- Relative standard deviation. aMean value of seven determinations. bBased on peak area. cBased on retention time | |
Accuracy and precision: To determine the accuracy and intra-day precision, pure ATN solutions at three different concentrations was analysed in seven replicates. The percent relative error, which is an index of accuracy, is <1.0% and indicates good accuracy. The relative standard deviation (<2%) at the 95% confidence level can be considered to be satisfactory. The inter-day precision was established by performing analyses over a period of five days on solutions prepared afresh each day (Table 1). The peak-area based and retention-time-based RSD values were <2.0 and <1%, respectively.
Linearity and range: Linearity was assessed in the range of 50 to 150% of the working level concentration including working level concentration. First and last level of linearity was carried out in six replicates and other levels in duplicates. The Linearity co-efficient of mean response of replicate determination plotted against respective concentration was found to be 0.9993. The percent y-intercept as obtained from the linearity data was less than 2%. The% RSD for peak area response of six replicates of first and last level was less than 2.0 and 1.0% for retention time.
Ruggedness (Intermediate Precision): Intermediate precision of six replicate determination of assay of a sample was analysed by different analyst with different instrument in different day after specifying the system suitability of the method. The % RSD of assay was less than 2.0% and the cumulative % RSD of assay of precision study and intermediate precision was also less than 2.0%.
Robustness: Robustness was done by altering deliberately two critical parameters by minor variation:
| • | Flow rate was changed to 1.1 mL min-1 |
| • | Column temperature changed to 32°C |
The % RSD for peak area response was less than 2.0 and 1.0% for retention time. The cumulative % RSD of assay of precision study and Robustness was also less than 2.0%.
Limit of quantitation and limit of detection: LOQ and LOD were established based on signal-to-noise ratio, performed by comparing measured signals from samples with known low concentrations of analyte with those of blank samples. LOQ and LOD were found to be 1.0 μg mL-1 and 0.4 μg mL-1, respectively.
Application: The results obtained are presented in Table 2 and found to be comparable, well with label claim. The results were also compared statistically with those obtained by a literature/reference method (Basavaiah and Chandrashekhar, 2006) by applying Student’s t-test for accuracy and F-test for precision. The proposed method was found to be superior as it has lower LOD (0.4 μg mL-1 against 0.625 μg mL-1) and lower Beer’s law limit suitable for trace level detection.
At the 95% confidence level, the calculated t- and F-values did not exceed the tabulated values (t = 2.77 and F = 6.39) suggesting that the proposed methods are as accurate and precise as the reference method.
The accuracy and validity of the proposed methods were further ascertained by performing recovery experiments. Pre-analysed tablet powder was spiked with pure ATN at three different levels and the total was found by the proposed methods. Each determination was repeated three times. The recovery of pure drug added was quantitative (Table 3) and revealed that co-formulated substances such as talc, starch, gelatin, gum acacia, calcium carbonate, calcium gluconate, calcium dihydrogen orthophosphate, sodium alginate and magnesium stearate did not interfere in the determination.
| Table 2: | Results of determination of Atenolol in formulations and statistical comparison with the reference method |
 | |
| dMean±SD, n = 5; #Marketed by: aNicolas pirmal; bZydus cedilla; cBlue cross. Tabulated t-value at 95% confidence level is 2.77; Tabulated F-value at 95% confidence level is 6.39 | |
| Table 3: | Recovery study |
 | |
| cMean±SD, n = 3; Marketed by: aNicolas pirmal; bZydus cedilla | |
| Table 4: | Comparison of proposed method with literature methods |
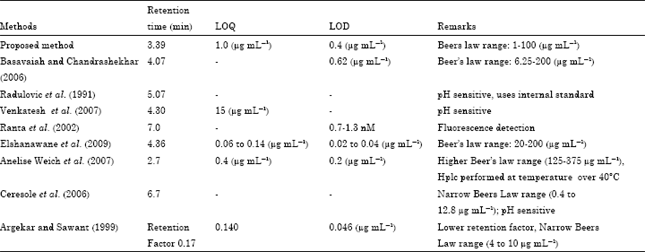 | |
The alternative HPLC method described here is compared with other previously published methodologies. However, these methods are pH sensitive and have relatively high retention times. (Basavaiah and Chandrashekhar, 2006; Radulovic et al., 1991; Venkatesh et al., 2007; Ranta et al., 2002; Elshanawane et al., 2009; Weich et al., 2007; Cersole et al., 2006; Argekar and Sawant, 1999) (Table 4).
CONCLUSION
The method is simple, rapid and convenient since it doesn’t need any internal standard. The method is characterized by shorter retention time and a wide linear dynamic range (1-100 μg mL-1) of concentration over which it is applicable. There was no interference from matrix sources. Through the statistical analysis and recovery studies (98.3-102.5%.) it is found that the proposed method can be effectively employed for quantification of atenolol in pharmaceutical formulations and real samples.
ACKNOWLEDGMENTS
The authors would like to thank the Management of Amrita Vishwa Vidyapeetham for their support during this research activity.
REFERENCES
- Elshanawane, A.A., S.M. Mostafa and M.S. Elgawish, 2009. Development and validation of a reversed-phase high-performance liquid chromatographic method for the simultaneous determination of amiloride hydrochloride, atenolol, hydrochlorothiazide and chlorthalidone in their combined mixtures. J. AOAC Int., 92: 404-409.
PubMed - Agarwal, Y.K., K. Raman, S. Rajput and S.K. Menon, 1992. Spectrophotometry determination of atenolol via hydroxamic acid formation. Anal. Lett., 25: 1503-1510.
Direct Link - Argekar, A.P. and J.G. Sawant, 1999. Simultaneous determination of atenolol and nitrendipine in pharmaceutical dosage forms by HPLC. J. Liquid Chromatogr. Related Technol., 22: 1571-1578.
CrossRef - Weich, A., D.C. de Olineira, J. de Melo, K. Goebel and C.M.B. Rolim, 2007. Validation of UV spectrophotometric and HPLC methods for quantitative determination of atenolol in pharmaceutical preparations. Lat. Am. J. Pharm., 26: 765-770.
Direct Link - Basavaiah, K., U. Chandrashekar and P. Nagegowda, 2006. Titrimetric, spectrophotometric and kinetic methods for the assay of atenolol using bromate-bromide and methyl orange. J. Serb. Chem. Soc., 71: 553-563.
Direct Link - Basavaiah, K. and U. Chandrashekhar, 2006. Determination of atenolol in pharmaceuticals by high performance liquid chromatography and spectrophotometry. Bulgarian Chem. Commun., 38: 104-111.
Direct Link - Coragem Briguenti, A.C. and P.S. Bonato, 2005. Quantitative Analysis of Beta-Blockers in pharmaceutical preparations by capillary electrophoresis. Drug Dev. Ind. Pharm., 31: 209-214.
Direct Link - Cersole, R., M.A. Moyano, M.T. Pizzorno and A.I. Segall, 2006. Validated reversed-phase HPLC method for the determination of atenolol in the presence of its major degradation product. J. Liquid Chromatogr. Related Technol., 29: 3009-3019.
CrossRef - El-Gindi, A., S. Emara and A. Moustafa, 2005. HPLC and chemometric-assisted pectrophotometric methods for simultaneous determination of atenolol, amiloride hydrochloride and chlorthalidone. Farmaco, 60: 269-278.
Direct Link - El-Gindy, A., S. Sallam and A.R. Abdel-Salam, 2008. HPLC method for the imultaneous determination of atenolol and chlorthalidone in human breast milk. J. Separation Sci., 31: 677-682.
Direct Link - Ferraro, M.C.F., P.M. Castellano and T.S. Kaufman, 2004. Chemometric determination of amiloride hydrochloride, atenolol, hydrochlorothiazide and timolol maleate insynthetic mixtures and pharmaceutical formulations. J. Pharm. Biomed. Anal., 34: 305-314.
CrossRef - Ferraro, M.C.F., P.M. Castellano and T.S. Kaufman, 2003. Chemometrics-assisted simultaneous determination of atenolol and chlorthalidone in synthetic binary mixtures and pharmaceutical dosage forms. Anal. Bioanal. Chem., 377: 1159-1164.
Direct Link - International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2005. ICH harmonised tripartite guideline, validation of analytical procedures: Text and methodology Q2(R 1). Complementary Guideline on Methodology Dated, 1996. Incorporated in November 2005, London. http://www.ich.org/LOB/media/MEDIA417.pdf.
- Jain, R. and C.L. Jain, 1991. Simultaneous microquantification of nifedipine and atenolol in multicomponent dosage forms using high performance liquid chromatography. Microchem. J., 44: 187-192.
CrossRef - Modamio, P., C.F. Lastra and E.L. Marino, 1998. Error structure for the HPLC analysis for atenolol, metoprolol and propranolol: A useful weighting method in parameter estimation. J. Pharm. Biomed. Anal., 17: 507-513.
CrossRef - Pawlak, Z. and B.J. Clark, 1992. The assay and resolution of the beta-blocker atenolol from its related impurities in a tablet pharmaceutical dosage form. J. Pharm. Biomed. Anal., 10: 329-334.
CrossRef - Phyramides, G., J.W. Robinson and S.W. Zito, 1995. The combined use of DSC and TGA for the thermal analysis of atenolol tablets. J. Pharm. Biomed. Anal., 13: 103-110.
CrossRef - Radulovic, D., L.T. Zivanovic, G. Velimirovic and D. Stevanovic, 1991. High-performance liquid chromatographic determination of atenolol in tablets. Anal. Lett., 24: 1813-1823.
Direct Link - Ruiz-Angel, M.J., R.D. Caballero, E.F. Simo-Alfonso and M.C. Garcia-Alvarez-Coque, 2002. Micellar liquid chromatography: Suitable technique for screening analysis. J. Chromatogr. A, 947: 31-45.
CrossRef - Sasa, S.I., I.M. Jalal and H.S. Khalil, 1988. Determination of atenolol combination with hydrochlorothiazide and chlorthalidone in tablet formulations by reverse-phase HPLC. J. Liquid Chromatogr., 11: 1673-1696.
Direct Link - Shafaati, A. and B.J. Clark, 1996. Development and validation of a capillary zone electrophoretic method for the determination of atenolol in presence of its related substances in bulk and tablet dosage form. J. Pharma. Biomed. Anal., 14: 1547-1554.
Direct Link - Sultan, S.M., 1992. Kinetic methods for the determination of atenolol in pharmaceutical preparations. Acta Pharm. Hung., 62: 311-317.
Direct Link - Ranta, V.P., E. Toropainen, A. Talvitie, S. Auriola and A. Urtti, 2002. Simultaneous determination of eight Beta-blockers by gradient high performance liquid chromatography with combined ultraviolet and fluorescence detection in corneal permeability studies in vitro. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci., 772: 81-87.
PubMed - Wadworth, A.N., D. Murdoch and R.N. Brodgen, 1991. Atenolol: A reappraisal of its pharmacological properties and therapeutic use in cardiovascular disorders. Drugs, 42: 468-510.
Direct Link - Wehner, W., 2000. Determination of atenolol and chlorthalidone during dissolution of tablets with UV multicomponent analysis. Pharmazie, 55: 543-544.
Direct Link - Wren, S.A. and P.J. Tchelitcheff, 2006. UPLC/MS for the identification of Beta-blockers. J. Pharm. Biomed. Anal., 40: 571-580.
Direct Link - Zakhari, N.A., S.M. Hassan and Y. El-Shabrawy, 1991. Colorimetric determination of beta-adrenergic blocking drugs with carbon disulphide and copper (I) ions. J. Pharm. Biomed. Anal., 9: 421-426.
Direct Link - Venkatesh, G., S. Ramanathan, S.M. Mansor, N.K. Nair, M. Abdul-Sattar, S.L. Croft and V. Navaratnam, 2007. Development and validation of RP-HPLC-UV method for simultaneous determination of buparvaquone, atenolol, propranolol, quinidine and verapamil: A tool for the standardization of rat in situ intestinal permeability studies. J. Pharm. Biomed. Anal., 43: 1546-1551.
CrossRefDirect Link