
Maryam S. Nuser
Department of Computer Information Systems, Yarmouk University (211-63), Irbid, Jordan
Trends in Bioinformatics
Year: 2010 | Volume: 3 | Issue: 1 | Page No.: 1-10
DOI: 10.3923/tb.2010.1.10







ABSTRACT
The aim of this study was to simulate an in vitro protocol that selects maximally mismatched DNA sequences (words). This simulation helps in estimating the protocol results and helps in expecting the difference in the results when running the protocol for very long times. It also assists in characterizing Non Cross-Hybridizing (NCH) DNA words. In addition, the simulation might be used to produce large libraries of Non Cross Hybridizing (NCH) DNA words in silico. Selection of words with good properties is important for reliable DNA based applications because words should hybridize as designed in order to provide correct results. These words can be used in solving large problems and in nano technology applications. Furthermore, several applications were proposed to show the applicability of the selection protocol in different disciplines. Analysis of the simulation of the protocol showed that the concentrations of the NCH words did increase in the population which shows an evidence of the ability of the protocol to produce NCH words. Most of the changes in concentrations occurred during the first few rounds of the protocol and not much change occurred after that. In addition, the results showed that only a small portion of the population will not crosshybridize. The results of the simulation model were verified experimentally with the lab results.
PDF Abstract XML References Citation
How to cite this article
Maryam S. Nuser, 2010. Simulation of an in vitro PCR Based Protocol with Applications. Trends in Bioinformatics, 3: 1-10.
DOI: 10.3923/tb.2010.1.10
URL: https://scialert.net/abstract/?doi=tb.2010.1.10
DOI: 10.3923/tb.2010.1.10
URL: https://scialert.net/abstract/?doi=tb.2010.1.10
INTRODUCTION
During the last decade, DNA computing (DNAC) has come to the fore with the goal of minimizing time, space complexities and the energy needed in computations. The main principal of DNAC is to utilize DNA molecules for computation and non-biological applications. Adleman (1994) uses DNA to solve the Hamilton Path Problem (HPP) which is an NP-complete problem. His solution required O (n) biological steps to solve a graph of size n with an exponential amount of DNA. Research continues to use the same methodology to solve NP-complete problems (Lipton, 1995; Braich et al., 2002). However, crosshybridization was one of several pitfalls that constraint DNA computation implementations. Crosshybridization, as shown in Fig. 1, represents the formation of double-stranded DNA hybrids, by complementary base pairing between two molecules that are not complementary in sequence. Large libraries of non-crosshybridization (NCH) DNA oligonucleotides helps DNAC to fulfill its potential through the large number of available molecules and parallelism of the reaction among them by allowing DNAC to scale to larger problems.
The main challenge in DNA computations and studies is the word design, which tries to find DNA libraries (NCH sets) with minimum crosshybridization (Deaton et al., 2002). Therefore, it is important to select good DNA words for computation because crosshybridization can lead to false positives.
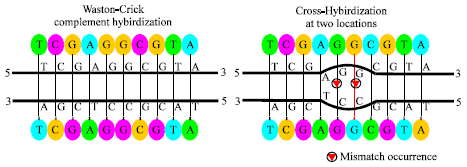 | |
| Fig. 1: | Cross hybridization |
Design tools should be developed to initiate libraries of NCH DNA words. These tools could be software based models of hybridization (Hartemink and Gifford, 1997; Rose et al., 1999) that depend on nearest neighbor model (Lucia, 1998), or the hamming distance (Frutos et al., 1997; Garzon et al., 1997), or it could be an actual protocol performed in the test tube (Deaton et al., 2003). In order to select the best DNA words for solving a specific problem, it is vital to understand hybridization among the DNA space. To find good encoding of problems, predict results of their protocols, prevent running in vitro protocols for longer times than needed and to determine the scope of problems that DNA can solve scientists should characterize the set of all possible hybridization on the DNA space.
Some researchers focus on designing DNA words on computers while others try to manufacture libraries of DNA words in the test tube. Deaton et al. (2006) designed and tested a protocol to select mismatched oligonucleotides for nanotechnology applications from a random starting material. Experimental results show that the selected short DNA words are independent and that there are about 10000 distinct words. Rather than investigating DNA words separately, Gao et al. (2009) used a pooling design algorithm to represent two new constructions whose efficiency ratio, i.e., the ratio between the number of tests and the number of items, is smaller than some of the existing constructions. On the other hand, Zhang et al. (2008) adopted an improved taboo search algorithm that finds NCH DNA words. The improved algorithm searches for good DNA words of a specific length and has the ability to overcome the disadvantage of taboo search by avoiding trapping into local optimization. Another algorithm that is based on the IWO algorithm is developed to optimize encoding words. The new algorithm was developed by defining the colonizing behavior of weeds to overcome the obstacles of the original IWO algorithm, which cannot be applied to discrete problems directly. The algorithm proposed an effective and convenient way for the user to design and select good DNA words in silicon that is suitable for DNA computing (Zhang et al., 2009).
Finding large sets of non-crosshybridizing DNA words is important because it provides more reliable computations since it decreases the error that might result from crosshybridization. Modeling and simulating a selected protocol for in vitro manufacturing of NCH DNA words can help indicate the trend of changes in concentrations of DNA words in the population and the concentrations of NCH words after running the protocol for several rounds. This study presents the simulation of a PCR based protocol in addition to applications of the protocol. It discusses the program used for the simulation, its results, the relation between these results and the DNA space and suggests ways where the protocol might be applicable.
MATERIALS AND METHODS
An in silico simulation of an in vitro selection protocol that selects maximally mismatched DNA words is done. The simulation was conducted at Yarmouk University in 2008. The following sections present the simulation and its results followed by suggested applications of the protocol.
Simulation of DNA Computations
DNA word design problem is considered a difficult computational problem as the generation of large sets of independent sequences is complex. Selection of words with good properties is important for reliable DNA based applications because words should hybridize as designed in order to provide correct results. One way to gain an insight into the characteristics of NCH DNA words is to analyze a selection protocol for a set of NCH words in vitro.
This research discusses a simulation model of an in vitro protocol that selects NCH oligonucleotides from a starting population of all possible words of a given length. The model is used to analyze the selection of NCH libraries from large populations of DNA words (DNA word problem) including DNA space of all possible words of a given length. Furthermore, the model is used to estimate the size of the NCH libraries, which determine the size of possible applications. Large libraries enable larger computations and nanostructures with more reliability and more precise control of biochemical operations.
Deaton et al. (2003) introduce a two steps PCR based in vitro protocol to produce NCH DNA. The first step constructed a starting population of words randomly while in the second step, the words with the desired property are isolated and then, amplified. The second step is repeated until either the specified number of sequences is reached, or there is no major modification in the population of sequences over time.
Starting with a random set of DNA molecules with primers attached at both ends of the molecules, a PCR amplification is done at a low temperature, which is then repeated until an amplification is detected (Deaton et al., 2003). After the first polymerization, all the selected words have their Watson-Crick complements in the test tube. Thus there was a concern that the selection pressure for independent Oligonucleotides, which is controlled by the melting temperature before polymerization, would be decreased by the addition of the Watson-Crick complement. As a result, one would expect that the concentration of independent sequences would not increase and that the presence of Watson-Crick complement would prevent the protocol from working as planned. However, given the vast number of sequences in the test tube, theoretically 420 for starting sequences of length 20 bases, the chances of words finding their complements is low, because in the protocol, the population of words are not allowed to reach equilibrium pairings. The primers on either end of the random sequences reinforce the tendency of pairs to be trapped in non-Watson- Crick pairings. In addition, Deaton et al. (2003) reported that the protocol actually does select for maximally mismatched sequences, but additional confirmation is needed. Besides, there was no information about the behavior of the protocol in the long run. Therefore, simulating the protocol provides predictions and more understanding of the behavior of the protocol.
Ideally, each DNA base should hybridize with its Watson-Crick (W-C) complement such that A binds with T and G binds with C and vice versa. To get the correct solution with the minimum possible error and in the minimum possible time, the DNA words should hybridize only as designed (Deaton et al., 1996; Brennenman and Condon, 2001). Otherwise, unwanted crosshybridizations will waste oligonucleotides because they do not contribute to the solution. Also, crosshybridization can lead to errors in the result; for example, there may be false negative or positive solutions. Thus, if DNA words can be found that have the minimum number of these unplanned crosshybridizations, then, the DNA computations will be more efficient and reliable. In addition, having large libraries of non-crosshybridizing (NCH) DNA oligonucleotides allows DNAC to scale to larger problems and will help DNAC to fulfill its potential through the large number of available molecules and the parallelism of the reactions among them.
Next, the model will be used to understand the protocol’s behavior and how does the concentration of good DNA words change. In addition, it will help building applications using that protocol.
The Simulation Model
In a previous simulation, DNA words were represented by their frequency in the test tube and a vector of pairwise hybridization energies with other words in the test tube (Nuser and Deaton, 2003). The energies were not real, instead they were calculated randomly. In this simulation model, a library of all possible DNA words of a specific length was generated. The energy was calculated according to a program that is written based on the NN model (Lucia, 1998) which represents the real free energy. If the energy (E), or interaction strength, between two words (wi, wj) was less than a threshold (t), then the words were considered to hybridize (h), otherwise they were independent (d) of each other. The relation (R) between two words wi and wj can be calculated as follows:
The free energy change for the hybridization will determine the concentrations of the product formed, whether it is a desired hybridization product, a mismatched hybridization, or hybridization with a shifted alignment. Where each different oligonucleotide duplex is given subscript i, ΔG (init) are the free energies for the possible W-C nearest neighbor stacking interactions, nij is the number of occurrences of each nearest neighbor j in each duplex i, ΔG (init) is the initiation free energy and ΔG (sym) equals -0.4 kcal mol-1 if duplex i is self complementary and zero otherwise (Lucia, 1998).
A C++ program was written to simulate the proposed model. The flow chart that shows the simulation algorithm is as shown in Fig. 2. The model was tested and found to be valid. The simulation results were compared to the experimental results from the lab. The simulation steps can be summarized as follows:
| • | Start |
| • | Assign a threshold value of t |
| • | Begin with a population of equal concentrations of DNA words |
| • | All possible words of length n |
| • | Calculate the probability of selection (ps): |
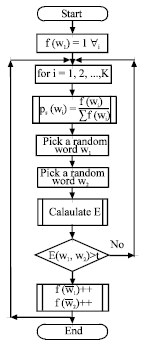 | |
| Fig. 2: | A flowchart that shows the simulation steps. f(wi) is the count of word wi, E is the energy between 2 words and |
| • | Proceed by randomly choosing two words according to their relative concentrations (number of occurrences) in the test tube |
| • | Calculate the pairwise free energy between the words E (wi, wj) |
| • | Compare the energy to a threshold |
| • | Simplify hybridization energetics |
| • | If E (wi, wj) is less than a threshold: |
| • | The two words are stable and hybridize with each other |
| • | Otherwise, if energy was equal or greater than the threshold: |
| • | Words do not hybridize | |
| • | The words are selected |
| • | Increment the number of their Watson-Crick complements in the population: |
| • | Simulate the copying by polymerase in the protocol |
| • | Recalculate the probability at each step: |
| • | To select the appropriate words | |
| • | Because the size of the population changed |
| • | Run the simulation for a set number of iterations |
| • | End |
RESULTS AND DISCUSSION
The program was run with a threshold value of -6.2. Different population sizes were tried. The sizes range from 1024 up to 65536. The main parameter was the concentration of NCH words in the population.
The results show that as the protocol starts to work, it selects NCH words and amplifies them in the population. With time, the concentration of NCH words increases in the population and therefore, the probability of selection for NCH words dominates that of CrossHybridizing words.
Running the protocol for a long time did not have a major change in the results. This is because the population, after a specific time, contains mostly NCH words and their selection probability is therefore much more than other words selection probability. Therefore, the probability of selection of words in the population remains almost constant. In addition, since the protocol selects words and based on their energy either amplifies them or not; words and their complements will be treated equally. This means that if in one round of the protocol, words wi and wj were selected and amplified, then if in another round words ![]() and
and ![]() were selected, then they should be amplified too. Therefore, the final population will have similar selection probability for words and their complements.
were selected, then they should be amplified too. Therefore, the final population will have similar selection probability for words and their complements.
For a population of 1024 DNA words, the simulation results show that there is a noticeable change in the concentration of words in the population for the first 1000 rounds. After that the change in the concentration starts to be minimized. Then after around 10,000 iteration, the concentrations appear to be almost constant.
Figure 3 shows the difference in concentrations of words (both CH and NCH words) while running the simulation model for different iterations. Figure 3A shows that initially the concentration of NCH DNA words is equal to the concentration of CH DNA words in the population. After several rounds of the protocol as shown in Fig. 3B, the concentration of NCH words becomes greater than the concentration of CH words. Then after more rounds the concentration of NCH words dominates other words in the population as shown in Fig. 3C.
Figure 4 shows the concentration (frequency) of words in a population of size 1024 over time. Initially, before running the protocol, the concentration is 1 for all words in the population, which is shown by the line with a diamond at the top. Then after 1000 iterations the concentration of some words increases up to 52 as shown by the lines with square at the top. These are the words that do not crosshybridize which the protocol amplifies over other words. After 10,000 iterations the concentrations of some limited (almost 20) words is increased and reach 224 as shown by the lines with triangle at the top.
The protocol was applied on a population of size 65536. After 100 iterations, the concentrations of NCH words increased and reached up to 5 copies. While after a 1000 iteration the changes in the concentrations appear more clearly and for some words reached up to 49 copies. Part of these results is shown in Fig. 5.
In spite of the ability of the simulation model to imitate the behavior of the in vitro protocol in amplifying NCH DNA words, there is a limitation to the simulation. The simulation model was implemented on a small scale compared to the length of real DNA words that are used in vitro.
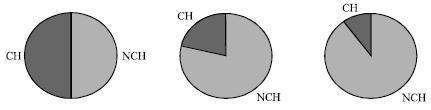 | |
| Fig. 3: | The concentration (frequency of words) of CH (cross hybridizing) words compared to the concentration of NCH words over time. (A) Time 10, (B) Time 1000, (C) Time 10000 |
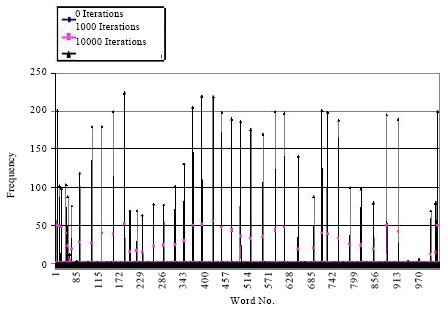 | |
| Fig. 4: | The frequency of DNA words at rounds = 0, 1000 and 10000 |
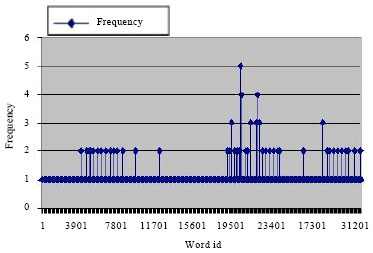 | |
| Fig. 5: | The frequency of DNA words after 100 iteration of the protocol |
This is because dealing with larger words increases the complexity of the algorithm which is NP-complete problem.
Applications of the Protocol
The simulated protocol can be used in different applications. The idea of the protocol is to amplify specific DNA words from the population. Therefore, several problems can be solved by representing the problem instances using DNA words and attach primers to these words. Using a specific primer, the wanted words can be amplified.
One example of an application of the protocol is to build an antivirus. Antivirus software discovers computer viruses mainly by comparing files with the content of a previously build dictionary of known viruses. If a match happened then the file is assumed to have a virus,otherwise the file is assumed to be clean.
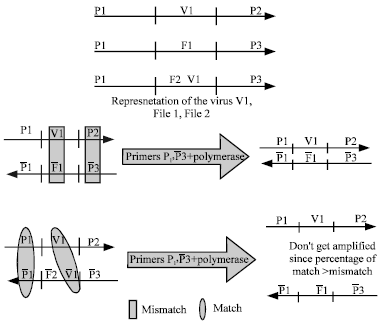 | |
| Fig. 6: | Applying the selection protocol on files to work as antivirus. Files with no viruses get amplified while files that have viruses do not |
A DNA antivirus can be build to isolate clean files and delete files that have viruses. This idea can be implemented by: first encoding the known virus and the files by specific DNA words with primers attached to them. Figure 6 shows the encoding of a virus named V1 with primers p1 and p2; a file named F1 with primers p1 and p3 that has no viruses and a file named F2 with primers p1 and p3 which has a virus V1.
In order for the antivirus to isolate files that do not have the virus V1, the second step would be to run the selection protocol with primers p1 and ![]() (the complement of p3). V1 will hybridize perfectly with
(the complement of p3). V1 will hybridize perfectly with ![]() and therefore the selection protocol will not select it for amplification. The same can be said about F1 and F2. On the contrary,
and therefore the selection protocol will not select it for amplification. The same can be said about F1 and F2. On the contrary, ![]() will have a mismatch with V1, which is shown by the rectangle in Fig. 6 and as a result will be amplified producing F1 which is the file that has no virus.
will have a mismatch with V1, which is shown by the rectangle in Fig. 6 and as a result will be amplified producing F1 which is the file that has no virus. ![]() which has the virus V1 will have a match (although not perfect but a great match) between V1,
which has the virus V1 will have a match (although not perfect but a great match) between V1,![]() and P1,
and P1, ![]() as shown by the ellipse in Fig. 6 and therefore will not be amplified. The single stranded DNA molecules will be eliminated with SS exonuclease. As a result, the DNA antivirus will amplify only files with no known viruses and delete (i. e., no amplification) files with viruses.
as shown by the ellipse in Fig. 6 and therefore will not be amplified. The single stranded DNA molecules will be eliminated with SS exonuclease. As a result, the DNA antivirus will amplify only files with no known viruses and delete (i. e., no amplification) files with viruses.
Another idea is to use the protocol to investigate human cells. The protocol can be used to increase the concentration of abnormal cells in the test tube in order to do more investigation and treatment search for these cells.
Another application in which the protocol can be used is spam detection. By amplifying messages (encoded by DNA words) that do not have unwanted words, the messages with high concentration can be extracted and delivered to the user.
CONCLUSION
This study presented a simulation model of a selection protocol for in vitro manufacturing of NCH DNA words. The simulation model was developed for an in vitro protocol to select independent oligonucleotides, though on a reduced scale. The primary goal of the simulation was to gain insight into whether independent pairs were actually selected based on the free energy and to observe the behavior of the concentrations in the test tube as the protocol is iterated. The simulation indicated the increase in the concentration of NCH words in the population. It also indicated that most of the changes in concentration occur after the first few steps of the protocol. In addition, the results generated from the simulation predicted an aspect about DNA spaces which is the little number of NCH DNA words compared to the space size. In addition, many applications were discussed to explore the computational capability of the selection protocol.
REFERENCES
- Adleman, L.M., 1994. Molecular computation of solutions to combinatorial problems. Science, 266: 1021-1024.
CrossRefPubMedDirect Link - Braich, R.S., N. Chelyapov, C. Johnson, P.W.K. Rothemund and L. Adleman, 2002. Solution of a 20-variable 3-sat problem on a DNA computer. Science 296: 499-502.
CrossRef - Brennenman, A. and A.E. Condon, 2001. Strand design for bio-molecular computation. Theor. Comput. Sci.
Direct Link - Deaton, R., J. Chen, H. Bi, M. Garzon, H. Rubin and D.H. Wood, 2003. A PCR-based protocol for in vitro selection of non-crosshybridizing oligonucleotides, in Hagiya and Ohuchi, DNA Computing. Proceedings of 8th International Workshop on DNA-Based Computers, (IWDNA-BC'03), Hokkaido University, Sapporo, Japan, pp: 196-204.
- Deaton, R., R.C. Murphy, M. Garzon, D.T. Franceschetti and S.E. Jr. Stevens, 1996. Good encodings for DNA-based solutions to combinatorial problems. Proceedings of the 2nd Annual Meeting on DNA Based Computers, June 10-12, American Mathematical Society, Princeton University, pp: 159-171.
Direct Link - Frutos, A.G., Q. Liu, A.J. Thiel, A.M.W. Sanner, A.E. Condon, L.M. Smith and R.M. Corn, 1997. Demonstration of a word design strategy for DNA computing on surfaces. Nucleic Acids Res., 25: 4748-4757.
PubMed - Gao, S., Z. Li, J. Yu, X. Gao and W. Wu, 2009. DNA library screening, pooling design and unitary spaces. Theor. Comput. Sci.
Direct Link - Nuser, M. and R. Deaton, 2003. Simulations of DNA computing with in vitro selection. GPEM., 4: 173-183.
CrossRef - Rose, J.A., R.J. Deaton, D.R. Franceschetti, M. Garzon and S.E. Stevens, 1999. A statistical mechanical treatment of error in the annealing biostep of DNA computation. Proceedings of the Genetic and Evolutionary Computation Conference, July 1999, AAAI, Morgan Kaufmann, San Francisco, Orlando, FL, USA., pp: 1829-1834.
- SantaLucia, J., 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA., 95: 1460-1465.
CrossRefDirect Link - Zhang, K., J. Xu, X. Geng, J. Xiao and L. Pan, 2008. Improved taboo search algorithm for designing DNA sequences. Prog. Nat. Sci., 18: 623-627.
CrossRef