
Shabrina N. Imana
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok, Indonesia
Eka G. Ningsih
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok, Indonesia
Usman S.F. Tambunan
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok, Indonesia
LiveDNA: 62.13001
Pakistan Journal of Biological Sciences
Year: 2020 | Volume: 23 | Issue: 4 | Page No.: 567-574







ABSTRACT
Background and Objective: Epidermal growth factor receptor (EGFR) is the biomarker for lung cancer in which the protein has the most active mutated genes in lung cancer patients. Peptides have pharmacological potential as drugs because of their bioactivity and accessibility. The research objective was to obtain peptide compounds drug candidates with good interaction and pharmacological properties that can act as an inhibitor for EGFR for lung cancer treatment by using in silico method. Materials and Methods: EGFR protein structure was obtained from Protein Data Bank and the peptide compounds were retrieved from PubChem. Optimization and energy minimization process were done to prepare the peptides for the simulation. Protein-Ligand Interaction Fingerprint (PLIF) was used to determine the pharmacophore features in the EGFR binding site. Both proteins and ligands underwent a virtual screening through rigid and flexible molecular docking simulation and the best ligands were subjected to a computational ADME-Tox properties prediction. Results: After screening through molecular docking simulation, nine best compounds were identified to have a good interaction with EGFR protein according to its binding energy and RMSD value. The compounds were identified to form hydrogen bond interactions with the macromolecule. Conclusion: Two peptide compounds (PubChem ID: 20832941 and 9805315) have been predicted as the best ligands with desired pharmacological properties for the inhibition of EGFR tyrosine kinase.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Shabrina N. Imana, Eka G. Ningsih and Usman S.F. Tambunan, 2020. In silico Identification of Peptide as Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Lung Cancer Treatment. Pakistan Journal of Biological Sciences, 23: 567-574.
DOI: 10.3923/pjbs.2020.567.574
URL: https://scialert.net/abstract/?doi=pjbs.2020.567.574
DOI: 10.3923/pjbs.2020.567.574
URL: https://scialert.net/abstract/?doi=pjbs.2020.567.574
INTRODUCTION
Cancer is an urgent global challenge because of the increased number of patients for the past few years. One type of cancer with the highest mortality rate around the world is lung cancer1. Lung cancer patients with non-small cell lung cancer (NSCLC) subtype is more common (around 85%) compared to small cell lung cancer (SCLC). NSCLC differed into several histological subtypes such as adenocarcinoma, large-cell carcinoma and squamous cell carcinoma. Epidermal growth factor receptor (EGFR) is the biomarker for lung cancer in which the protein has the most active mutated genes in patients with NSCLC2. EGFR is a transmembrane receptor tyrosine kinase protein that is expressed in some normal neurogenic mesenchymal and epithelial tissue. EGFR plays an essential role in the process of controlling cell growth, cell resistance and cell apoptosis3.
Gefitinib, an EGFR receptor tyrosine kinase inhibitors (TKIs) is an oral-administrated small molecule inhibitor that already approved by Food and Drug Administration (FDA), plays as a first or second-line treatment for advanced adenocarcinoma3,4. EGFR mutation genes are divided into two groups, namely EGFR mayor gene mutations (insertion/deletion exon 19 and L858R) and minor EGFR mutation genes (G719X and L861Q). Intracellular mutations occur and may impact a drug efficacy because it confers an increased affinity for these drugs in EGFR protein. This has been reported in several patients with NSCLC acquired resistance to gefitinib5,6.
In this research, in silico approach was used to obtain peptide compounds as EGFR tyrosine kinase inhibitor, hence become a promising drug in lung cancer. Peptides have pharmacological potential as drugs because of their bioactivity, low toxicity (related to its constituent amino acids), high diversification, high potential and selectivity, high efficiency, safe and tolerant, not bioaccumulative and easy to be synthesized and their accessibility are considered as a high potential to meet the desires of new drugs7. Furthermore, the development of more efficient and economic peptide synthesis and the improvement of peptide purification systems have been essential for the revival of the peptide field in recent decades8.
Discovering various target proteins and potential inhibitor to be developed as new drugs could be facilitated by combining genomic and proteomic studies with computational sciences9. In silico method or computer-aided drug discovery and development is a rapid developing field that grows to reduce the cost and improve time efficiency in drug development10. The molecular docking simulation is a method that has been developed to analyze interactions, affinity and stability of a ligand targeting other biomolecules11. Hence, the use of in silico method can eliminate the possible undesired drug candidate.
This study was conducted to obtain the information of the potential peptide compounds as a novel inhibitor for EGFR tyrosine kinase through in silico pharmacological test and molecular docking simulation method.
MATERIALS AND METHODS
This study was conducted at Bioinformatics Laboratory, Department of Chemistry, Faculty of Mathematics and Natural Sciences Universitas Indonesia from July, 2019-November, 2019. The method used in this study was based on the previous publication12.
Protein preparation: The three-dimensional (3D) structure of EGFR proteins were acquired from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). Each crystal structures have one unique ligand. Four proteins have PDB ID: 2ITW (2.88 Å), 2ITX (2.98 Å), 2ITY (3.42 Å) and 2J6 M (3.1 Å) with ITQ, ANP, IRE and AEE (Fig. 1) as unique ligands, respectively13. All protein structures were prepared using force field Amber10: EHT in R-Field solvation by utilizing Molecular Operating Environment (MOE) 2014.09 software. The process was initiated with removing water molecules and continued to LigX stage for optimizing all structures. The prepared proteins were saved in MOE format (moe).
Pharmacophore designing: In MOE software, 3D pharmacophore models were determined through the stage of superpose and protein-ligand interaction fingerprints (PLIF) based on the ligand-based pharmacophore approach. The pharmacophore models were saved in pH4 format (ph4) for use in the next simulation.
Standard ligand preparation: Four standard ligands (Fig. 1) have consisted of all unique ligands from four proteins. The ligands were acquired from ChemSpider and saved in MDB format (mdb). The preparation process was conducted with MOE 2014.09 software by applying force field: MMFF94x and Gas-Phase as solvation. The process started with ‘Wash’ in default parameter and ‘Energy Minimization’ with RMS gradient of 0.001 kcal mol–1 Å.
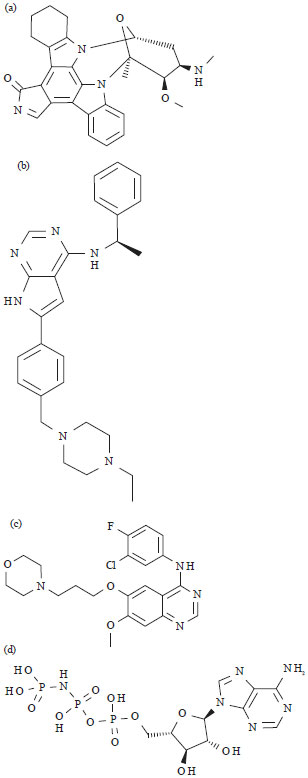 | |
| Fig. 1(a-d): | Standard ligand, (a) 1,2,3,4-tetrahydrogen staurosporine (ITQ), (b) 6-{4-[4-ethylpiperazine-1-Y1)methyl]phenyl}-N-[(1r)-1-phenylethyl]-7h-pyrrolo[2,3-D]pyrimidin-4-amine (AEE788), (c) Gefitinib (IRE) and (d) Phosphoaminophosphonic acid-adenylate ester (ANP) |
Construction of peptide database: Prior to database preparation, 8,629 peptide compounds were obtained from PubChem database. These peptide compounds were screened by using OSIRIS DataWarrior software. The unwanted compounds that showed the toxicity properties like mutagenic, tumorigenic, reproductive effect and irritant must be eliminated. About 3,258 molecules were prepared through similar parameters and protocols as the standard ligands. Then, the peptide database was saved on mdb format.
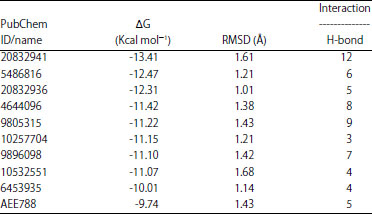
Molecular docking: Molecular docking simulation was performed on MOE software with force field AMBER10: EHT and R-Field solvation. Both standard ligands and peptide compounds were performed by using pharmacophore-based molecular docking simulation with two main protocols, such as rigid docking (30-1 and 100-1) and flexible docking (100-1). Then, the result was filtered based on the G binding value and RMSD value <2 Å. Nine ligands molecular interaction was analyzed and determined as the two best ligands.
ADME-Tox properties: After several screenings, the potential ligand from molecular docking simulation went through the initial pharmacological properties using SwissADME. This prediction applied to find the health effect from the potential ligands in the human organ.
RESULTS
Pharmacophore features generations: Several proteins were utilized through PLIF method. This method was conducted to obtaining the pharmacophore features in binding site of the protein. All protein and their ligands are aligned in MOE software (Fig. 2a) to predict the pharmacophore features. The 3D visualization of the protein shown that there are two pharmacophore features in the binding site, such as Acc and ML and Don (Fig. 2b).
Molecular docking simulation: Before the molecular docking simulations method, peptide ligands were conducted through the initial screening using OSIRIS DataWarrior Software to screen all unwanted compounds based on their toxicity properties. Within this method, only 3,258 peptide compounds remained for the molecular docking simulations.
In the molecular docking simulations, the two main protocols are conducted to obtain the best ligand based on their lower G binding, RMSD <2 Å and hydrogen bond interaction lower than standards (Table 1). Four standard ligands (Fig. 1) also underwent docking simulations using rigid and flexible docking protocols. Only AEE788 (Fig. 1b) standard compound screened up to the final stage. The number of ligands that predicted trough these several methods described in Fig. 3.
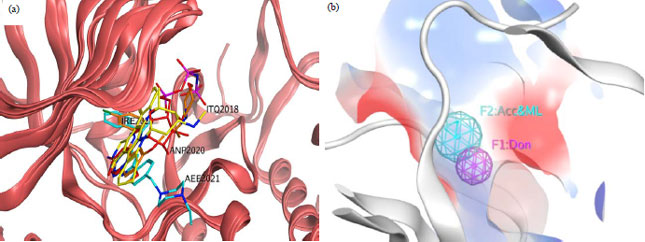 | |
| Fig. 2(a-b): | (a) 3D structure from all protein and its ligand during the PLIF method, the specified color of the ligand standard explains respectively, blue color refers to ligand AEE 788, Pink color explains ligand ITQ, orange color represents IRE, the red color represents ANP ligand and (b) Pharmacophore point Acc and ML (Turquoise) and Don (Purple). The red and blue in the pocket binding refers to the lipophilicity of the binding site in the protein |
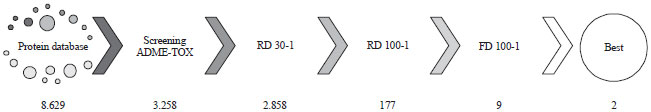 | |
| Fig. 3: | Number of ligands obtained in every step in molecular docking simulation and ADME-Tox prediction using MOE 2014.0901 and OSIRIS DataWarrior |
| Table 1: | dG binding, RMSD and H-bond interaction value of the best ligand and standard, from induced-fit docking simulation |
 | |
The visualization of 3D and 2D molecular interaction of best peptide ligands shown in Fig. 4. Ligand 20832941 (Fig. 4a) has interacted through seven hydrogen bond interactions with Ala722, Glu762, Gln791, Met793, Asn842, Asp855 and Gly857. Additionally, the Ala722, Leu792 and Pro877 have also had an interaction with Ligand 20832941 through aromatic π-π interaction. About twenty-seven amino acid residues in the EGFR protein have interaction with the 20832941. Another best ligand namely ligand 9805315 (Fig. 4b) was interacted with twenty amino acid residues of the EGFR protein binding site, with six hydrogen bond interactions (Ala722, Glu762, Gln791, Met793, Asn842, Asp855 and Gly857). From the both best ligand, there are four residues (Asp 855, Glu 762, Gln 791 and Met 793) that remain important, which has forms hydrogen bond interactions.
ADME-Tox prediction: The ADME-Tox Predictions were conducted using SWISS-ADME. With this method, several information such as gastrointestinal absorption (GI), cytochrome (CYP) inhibitor, drug-likeness properties such as bioavailability and medicinal chemistry (PAINS and synthetic accessibility) were gained and summarized in Table 2. Based on the data, it found that the AEE 788 standard ligand was inhibited several cytochromes enzymes such as CYP 2C19, CYP 2C9, CYP 2D6 and CYP 3A4.
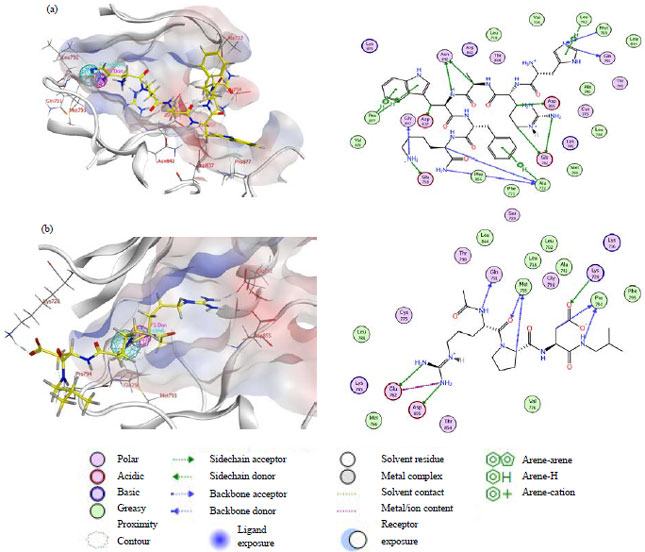 | |
| Fig. 4(a-b): | Two candidates of best ligand with PubChem ID, (a) 20832941 and (b) 9805315 in 3D and 2D visualization interaction |
In the 2D visualization for each ligand, the green dashed line explains sidechain acceptor/donor and blue dashed line represent backbone acceptor/donor, every line indicates the interaction between ligand and protein for green dashed line with arene-H (2D visualization in Fig. 4a) indicates the interaction between hydrogen and aromatic molecules in the structure | |
| Table 2: | ADME-Tox, drug-likeness and medical chemistry properties using SwissADME |
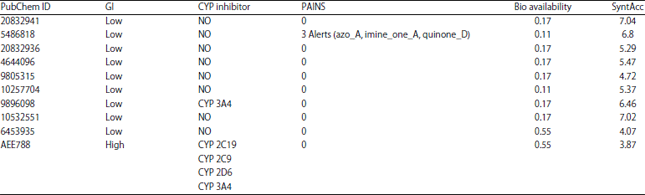 | |
DISCUSSION
In this study, all proteins (PDB ID: 2ITW, 2ITX, 2J6M, 2ITY) were prepared before docking simulation using MOE 2014.09. The water molecules were eliminated because the solvation effect is not taken into account in the molecular docking simulation14. All proteins used in this research were retrieved through X-ray crystallography, which usually lacks in hydrogen atoms. It is important to have a full atom on protein structures because it will influence the molecular mechanics, dynamics and electrostatic calculations involved in molecular docking simulation14,15. The Amber10: EHT was chosen as a forcefield in this protein preparation because it is suitable for protein. The Amber10:EHT is a force field that combines Amber10 with Extended Hückel Theory (EHT) bonded parameters in order to include electronic effects16,17.
After prepared the protein, peptide ligands and standard compounds also were utilized into the preparation mechanism. These molecules underwent ADME-Tox screening and energy minimization process as well described in the material and methods. Energy minimization is a necessary part of pre-docking preparation because it helps to maintain ligand into the lower ΔG value. The lower ΔG will affect the conformation of the ligand and it considered closed with the biological system.
The pharmacophore's point is necessary to identify from EGFR protein. The pharmacophore point is an important point that shows specific biological activity which is indicated by aligning the target protein of the drug and also the ligand which is known to be able to carry out the inhibitory activity of the protein18,19. In this study, the determination of pharmacophore points was performed using the PLIF method. Through the PLIF method, the fingerprints covered in a standardized quantitative score represent the similarities between the docking pose interaction profile and that of the protein-ligand reference complex20.
Protein with PDB ID: 2ITW with a resolution of 2.88 Å with 1,2,3,4-tetrahydrogen staurosporine (ITQ) as their inhibitor in their binding site was chosen for docking with consideration of the low protein resolution compared to other proteins. Protein with the minimum resolution is preferable to ensure a better quality of protein structure. The binding site of the EGFR protein consists of Leu718, Ala743, Gly796, Cys797, Asp800, Thr854, Arg841, Leu844, Thr854, Arg841, Asp855, Thr790, Gln791, Leu792, Met793, Pro794 and Phe795 amino acid residues. Determining the binding site that has potential pharmacological properties must be done. Consequently, targeting the identified binding site known to control EGFR protein stabilization. This method was preferable to identify binding sites rather than using an unknown binding site for a particular protein that needs more validation21.
ADME-Tox prediction for nine peptide compounds and one standard ligand were shown in Table 2. Comparing peptide compounds and standard ligand in ADME-Tox prediction is necessary to identify the mechanism for drug administration. Table 2 shows the GI absorption and bioavailability score predict the compound could act as an oral drug in drug administration. The high score of GI absorption and bioavailability in standard ligand determine this drug possible to act as an oral drug. Compared with the standard ligand, nine peptides have low scores in GI absorption and bioavailability. It means, the peptides have better performance if administrated by intravenous injection. Synthetic accessibility value is a score based on the fragmental analysis of the compound structure by the assumption of their size and complexity. The synthetic accessibility score range is between 1 (easy to synthesis) and 10 (difficult to synthesis).
AEE788 as standard was identified to inhibit several cytochrome P450 enzymes such as 2C19, 3A4, 2C9 and 2D6. In the previous study, AEE788 could act as an inhibitor for Vascular Endothelial Growth Factor (VEGF) and the Epidermal Growth Factor Receptor (EGFR). VEGF and EGFR play important roles in tumor growth and progression22,23. The utilization of AEE788 as a drug has been obtained to cure Glioblastoma patients. In the first phase of the clinical trial, several side effects were observed such as diarrhea, rashes and hepatotoxicity24. Deng et al.25 suspect that the metabolism of the drug might be liver size-dependent in the patient with 1 week of treatment. It might accumulate in the early postoperative phase and may represent an overdose in the patient with full-size liver25. As a result, the usage of AEE788 as a drug must be postponed for further research.
In this research, two ligands have been decided to be the best ligand which acts as a tyrosine kinase inhibitor. Based on RMSD value <2.0 Å, ΔG binding value lower than standard ligand and their H-bond (Table 1), two best ligand candidates (PubChem ID: 20832941 and ligand 9805315) have been chosen. Even though the bioavailability for ligand with PubChem ID: 6453935 considered high rather than these two compounds. But in protein-ligand interactions, hydrogen bonding contributed much to protein-ligand formation stability, build a scoring function for protein-ligand interaction prediction and developed a hydration penalty score for protein-ligand interactions26.
For further research, the molecular dynamics simulations approach is necessary to determine the stability of the two best ligands. It is expected that by knowing the stability of the two best ligands can find out the optimal conditions of the ligand that will be used as a drug before conducting in vitro and in vivo studies.
CONCLUSION
The initial screening and molecular docking simulation studies were performed to determine the most potential peptide compounds as an inhibitor of EGFR tyrosine kinase. About, 8,629 peptide compounds were screened towards these methods. From, the result of this research, two peptide ligands, namely ligand 20832941 and ligand 9805315 in PubChem were revealed as the most potent drug candidates for EGFR tyrosine kinase for the lung cancer treatment determining from their ΔG binding value, RMSD value, molecular interactions and ADME-Tox properties. Hence, the molecular dynamics studies should be conducted afterward to predict the ligand-protein complex stability which has been formed during the docking simulation.
SIGNIFICANCE STATEMENT
This research study is to develop new drug candidates for tyrosine kinase inhibitors for lung cancer patients. New drug candidates need to conquer the negative effect of the previous drug especially when the previous drug was had a negative effect on the cytochrome P450 This study will provide additional insight on the lung cancer drug discovery and development and further studies are needed to validate these compounds stability and efficacy.
ACKNOWLEDGMENT
This research is financially supported by Hibah Publikasi Internasional Terindeks 9 (PIT 9) Universitas Indonesia Tahun Anggaran 2019 No: NKB-0036/UN2.R3.1/HKP.05.00/2019. All authors were responsible for the writing of the manuscript. Hence, there is no conflict of interest regarding this project.
REFERENCES
- Zappa, C. and S.A. Mousa, 2016. Non-small cell lung cancer: Current treatment and future advances. Translat. Lung Cancer Res., 5: 288-300.
CrossRefPubMedDirect Link - Da Cunha Santos, G., F.A. Shepherd and M.S. Tsao, 2011. EGFR mutations and lung cancer. Annu. Rev. Pathol.: Mech. Dis., 6: 49-69.
CrossRefDirect Link - Patel, N., P. Wu and H. Zhang, 2017. Comparison of gefitinib as first- and second-line therapy for advanced lung adenocarcinoma patients with positive exon 21 or 19 del epidermal growth factor receptor mutation. Cancer Manage. Res., 9: 243-248.
CrossRefPubMedDirect Link - Nishimura, Y., 2018. Losmapimod: A novel clinical drug to overcome gefitinib-resistance. EBioMedicine, 28: 2-3.
CrossRefDirect Link - Paez, J.G., P.A. Janne, J.C. Lee, S. Tracy and H. Greulich et al., 2004. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science, 304: 1497-1500.
CrossRefDirect Link - Galdiero, S. and P.A.C. Gomes, 2017. Peptide-based drugs and drug delivery systems. Molecules, Vol. 22, No. 12.
CrossRefDirect Link - Di, L., 2015. Strategic approaches to optimizing peptide ADME properties. AAPS J., 17: 134-143.
CrossRefDirect Link - Tambunan, U.S.F., N. Apriyanti, A.A. Parikesit, W. Chua and K. Wuryani, 2011. Computational design of disulfide cyclic peptide as potential inhibitor of complex NS2B-NS3 dengue virus protease. Afr. J. Biotechnol., 10: 12281-12290.
Direct Link - Kapetanovic, I.M., 2008. Computer-Aided Drug Discovery and Development (CADDD): In silico-chemico-biological approach. Chemico-Biol. Interact., 171: 165-176.
CrossRefDirect Link - De Ruyck, J., G. Brysbaert, R. Blossey and M.F. Lensink, 2016. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Applic. Bioinform. Chem., 9: 1-11.
CrossRefPubMedDirect Link - Nasution, M.A.F., E.P. Toepak, A.H. Alkaff and U.S.F. Tambunan, 2018. Flexible docking-based molecular dynamics simulation of natural product compounds and Ebola virus nucleocapsid (EBOV NP): A computational approach to discover new drug for combating Ebola. BMC Bioinform., Vol. 19.
CrossRefDirect Link - Yun, C.H., T.J. Boggon, Y. Li, M.S. Woo, H. Greulich, M. Meyerson and M.J. Eck, 2007. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell, 11: 217-227.
CrossRefDirect Link - Tambunan, U.S.F., A.H. Alkaff, M.A.F. Nasution, A.A. Parikesit and D. Kerami, 2017. Screening of commercial cyclic peptide conjugated to HIV-1 Tat peptide as inhibitor of N-terminal heptad repeat glycoprotein-2 ectodomain Ebola virus through in silico analysis. J. Mol. Graph. Model., 74: 366-378.
CrossRefDirect Link - Oksanen, E., J.C.H. Chen and S.Z. Fisher, 2017. Neutron crystallography for the study of hydrogen bonds in macromolecules. Molecules, Vol. 22, No. 4.
CrossRefDirect Link - Li, M.J., G.Z. Wu, Q. Kaas, T. Jiang and R.L. Yu, 2017. Development of efficient docking strategies and structure-activity relationship study of the c-Met type II inhibitors. J. Mol. Graph. Model., 75: 241-249.
CrossRefDirect Link - Thapa, B., D. Beckett, J. Erickson and K. Raghavachari, 2018. Theoretical study of protein-ligand interactions using the molecules-in-molecules fragmentation-based method. J. Chem. Theory Comput., 14: 5143-5155.
CrossRefDirect Link - Schuster, D., 2019. Fingerprints and Pharmacophores. In: Encyclopedia of Bioinformatics and Computational Biology, Ranganathan, S., M. Gribskov, K. Nakai and C. Schonbach (Eds.). Vol. 2, Elsevier Inc., New York, USA., ISBN: 978-0-12-811432-2, pp: 619-627.
Direct Link - Chandrasekaran, B., N. Agrawal and S. Kaushik, 2019. Pharmacophore Development. In: Encyclopedia of Bioinformatics and Computational Biology, Ranganathan, S., M. Gribskov, K. Nakai and C. Schonbach (Eds.). Vol. 2, Elsevier Inc., New York, USA., ISBN: 978-0-12-811432-2, pp: 677-687.
Direct Link - Da, C. and D. Kireev, 2014. Structural Protein-Ligand Interaction Fingerprints (SPLIF) for structure-based virtual screening: Method and benchmark study. J. Chem. Inform. Model., 54: 2555-2561.
CrossRefDirect Link - Meng, X.Y., H.X. Zhang, M. Mezei and M. Cui, 2011. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des., 7: 146-157.
CrossRefDirect Link - Tabernero, J., 2007. The role of VEGF and EGFR inhibition: Implications for combining anti–VEGF and anti–EGFR agents. Mol. Cancer Res., 5: 203-220.
CrossRefDirect Link - Traxler, P., P.R. Allegrini, R. Brandt, J. Brueggen and R. Cozens et al., 2004. AEE788: A dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res., 64: 4931-4941.
CrossRefDirect Link - Reardon, D.A., C.A. Conrad, T. Cloughesy, M.D. Prados and H.S. Friedman et al., 2012. Phase I study of AEE788, a novel multitarget inhibitor of ErbB- and VEGF-receptor-family tyrosine kinases, in recurrent glioblastoma patients. Cancer Chemother. Pharmacol., 69: 1507-1518.
CrossRefDirect Link - Deng, M., H. Huang, O. Dirsch and U. Dahmen, 2010. Effect and risk of AEE788, a dual tyrosine kinase inhibitor, on regeneration in a rat liver resection model. Eur. Surg. Res., 44: 82-95.
CrossRefDirect Link - Zhou, W., H. Yan and Q. Hao, 2012. Analysis of surface structures of hydrogen bonding in protein-ligand interactions using the alpha shape model. Chem. Phys. Lett., 545: 125-131.
CrossRefDirect Link