
Wuyi Liu
Department of Biology Sciences, Faculty of Biological and Food Engineering, Fuyang Normal University, Fuyang City, China
Fengmei Li
Department of Biology Sciences, Faculty of Biological and Food Engineering, Fuyang Normal University, Fuyang City, China
Pakistan Journal of Biological Sciences
Year: 2015 | Volume: 18 | Issue: 4 | Page No.: 149-165







ABSTRACT
Xenopus laevis is a long established model organism for developmental, behavioral and neurological studies. Herein, an updated genome-wide survey was conducted using the ongoing genome project of Xenopus laevis and 106 non-redundant Basic Helix-Loop-Helix (bHLH) genes were identified in the Xenopus laevis genome databases. Gene Ontology (GO) enrichment statistics showed 51 significant GO annotations of biological processes and molecular functions and 5 significant KEGG pathways and a number of Xenopus laevis bHLH genes play significant role in specific development or special physiology processes like the development processes of muscle and eye and other organs. Furthermore, each sub-group of the bHLH family has its special gene functions except for the common GO term categories. Molecular phylogenetic analyses revealed that among these identified bHLH proteins, 105 sequences could classified into 39 families with 46, 25, 10, 5, 16 and 3 members in the corresponding high-order groups A, B, C, D, E and F, respectively with an addition bHLH member categorized as an orphan. The present study provides much useful information for further researches on Xenopus laevis.
PDF Abstract XML References Citation
Received: June 03, 2015;
Accepted: July 23, 2015;
Published: August 05, 2015
How to cite this article
Wuyi Liu and Fengmei Li, 2015. Identification and Bioinformatics Analyses of the Basic Helix-loop-helix Transcription Factors in Xenopus laevis. Pakistan Journal of Biological Sciences, 18: 149-165.
DOI: 10.3923/pjbs.2015.149.165
URL: https://scialert.net/abstract/?doi=pjbs.2015.149.165
DOI: 10.3923/pjbs.2015.149.165
URL: https://scialert.net/abstract/?doi=pjbs.2015.149.165
INTRODUCTION
Transcription Factors (TFs) are frequently identified and classified into families or subfamilies based on the sequence similarity or comparability of DNA-binding domains which are highly conserved among species. Some TF families are common to most eukaryotic organisms, while others TF families are specific to particular taxonomic groups (Luscombe et al., 2000; Riechmann et al., 2000). The Basic Helix-Loop-Helix (bHLH) transcription factors constitute one of the largest families of functionally important proteins and were believed to be key regulators in cell proliferation and differentiation, cell lineage determination, the formation of muscle, neurons, gut and blood, sex determination, as well as other essential developmental and genetic processes (Atchley and Fitch, 1997; Massari and Murre, 2000; Ledent and Vervoort, 2001; Jones, 2004). Due to the interaction networks and important functions that they display in various organisms, bHLH TFs have been the subject of many researches designed to identify and characterize their functions and interactions and classification informations. The first identification of bHLH TFs was reported by Murre et al. (1989) who focused on the murine factors E12 and E47 and then an increasing number of bHLH TFs has been characterized by scholars, particularly in recent years. The studies of animal bHLH proteins have led to the definition of six major functional and evolutionary lineages or groups (A-F) that can be further subdivided into smaller orthologous families named after the first discovered or best-known member (Atchley and Fitch, 1997; Ledent and Vervoort, 2001; Ledent et al., 2002; Jones, 2004; Skinner et al., 2010). Groups A and B bHLH proteins bind to E boxes (CANNTG), in which group A binds to sequences CACCTG or CAGCTG and group B binds to sequences CACGTG or CATGTTG. Group C proteins are complex molecules with one or two PAS domains following the basic helix-loop-helix motif. They bind the core sequences of ACGTG and/or GCGTG. Group D proteins lack a basic domain and form inactive heterodimers with group A bHLH proteins. Group E proteins bind preferentially to core sequences typical of N boxes (CACGCG or CACGAG). Group F lack the basic domain part and are characterized by the presence of an additional domain for DNA binding and dimerization, the COE domain. The bHLH TFs share a common bHLH motif or domain of 60 amino acids or so which holds a basic region and two helices separated by a loop (HLH) region of variable length (Massari and Murre, 2000; Ledent and Vervoort, 2001). The basic region is a DNA-binding domain. The amphipathic α-helices of two bHLH proteins can interact can interact with each other and the HLH domain promotes dimerization, allowing the formation of homodimeric or heterodimeric protein complexes between different members (Ledent and Vervoort, 2001). Atchley et al. (1999) computed and deduced a predictive motif for the bHLH domains based on 242 bHLH proteins, in which 19 conserved sites were found within the bHLH domain (Atchley et al., 1999). Their works showed that a sequence identified with less than 8 mismatches to the predictive motif was possibly a bHLH protein (Atchley and Fitch, 1997; Atchley et al., 1999; Atchley et al., 2000) and other researchers found that a sequence with nine mismatches might be a potential bHLH protein as well (Toledo-Ortiz et al., 2003).
With the genome resources of interested organisms being available, it would be desirable to have a more refined classification scheme of various types of bHLH motifs, as well as a better understanding of their functions and evolutionary implications within and/or among species. Recently, more and more bHLH genes has been identified and bHLH TF families have been analyzed in many organisms whose genome drafts have been available (Ledent et al., 2002; Toledo-Ortiz et al., 2003; Buck and Atchley, 2003; Heim et al., 2003; Li et al., 2006a, b; Simionato et al., 2007; Stevens et al., 2008; Wang et al., 2007, 2008, 2009; Zheng et al., 2009; Pires and Dolan, 2010; Carretero-Paulet et al., 2010; Liu and Zhao, 2010, 2011; Liu et al., 2012, 2013; Liu and Chen, 2013). However, the family of bHLH TFs has not yet been studied and characterized in Xenopus laevis. The Xenopus laevis and its congener Xenopus tropicalis are established model organisms for biological researches in development, behavior and neurology (Carruthers and Stemple, 2006; Bowes et al., 2008). The draft of Xenopus tropicalis genome assembly was recently accomplished by American scientists at the Lawrence Berkeley National Laboratory (Hellsten et al., 2010) but the Xenopus laevis ongoing genome project has not been accomplished because of its genomic complex. Our interest focuses on the identification and functional and evolutionary analysis of the bHLH TFs in Xenopus laevis. In a previous work (Liu, 2011), the preliminary survey identified 98 bHLH TFs in Xenopus laevis but it was proved to be incomplete. Therefore, we conducted an updated genome-wide survey here using the ongoing genome project database of Xenopus laevis and identified 106 bHLH sequences in its genome. In the study, we used both the predictive motif developed by Atchley et al. (1999) and the 45 representative bHLH domains defined by Ledent et al. (2002) and Simionato et al. (2007) to do similarity searches using BLAST algorithm in the Xenopus laevis genomic database and finally identified 106 bHLH proteins. We also did similarity searches using BLAST algorithm with 105 Xenopus tropicalis bHLH domains (Liu and Chen, 2013) (Table 1). Next, we made phylogenetic analyses of the Xenopus laevis bHLH factors with 118 human bHLH proteins (Simionato et al., 2007) which allowed us to define the Xenopus laevis bHLH orthologous genes. We further reported the result of gene function enrichment of the Xenopus laevis bHLH proteins using GO annotations.
MATERIALS AND METHODS
Similarity searches using BLAST algorithm and retrieval of bHLH proteins: We initially followed the criteria developed by Atchley et al. (1999) to define a bHLH protein and retrieved 8 bHLH sequences in primary searches based on the protein domain consensus sequences predicted by Atchley et al. (1999). The predictive motif is:
‘++X(3–6)E+XRX(3)αNX(2)ΦX(2)L+X(5–22)+X(2)
KX(2)δLX(2) AδXYαX(2)L’
where, + is K, R, α is I, L, V, Φ is F, I, L, δ is I, V, T, E, R, K, A and Y are as defined, X is any residue, X(I) is any i residues and X(i-j) is i to j of any residues.
Then, we used both these initially retrieved bHLH sequences and the 45 representative bHLH domains to make BLASTP and TBLASTN and PSI-BLAST searches of bHLH domains. Each sequence was then used to perform multiple searches against the non-redundant databases of Xenopus laevis built by NCBI (http://www.ncbi.nlm.nih.gov/ genome/seq/BlastGen/BlastGen.cgi?taxid = 8355) and the Xenopus laevis Genome Project databases (http://xenopus. lab.nig.ac.jp/). Stringency was set to E<10 in order to obtain all bHLH-related sequences for later examination. With TBLASTN against the Xenopus laevis database, we obtained all putative bHLH proteins that had more than 10 conserved amino acids among the 19 residues (Toledo-Ortiz et al., 2003). Each sequence was used to perform second and third TBLASTN and/or PSI-BLAST searches against the Xenopus laevis genomic database. We also checked the Xenbase Release v3.3 (http://www.xenbase.org) (Bowes et al., 2010; Karpinka et al., 2015) used the previous obtained 105 Xenopus tropicalis bHLH domains (Table 1) to make BLASTP and TBLASTN and PSI-BLAST searches (Liu and Chen, 2013).
| Table 1: | Information of the 105 Xenopus tropicalis basic helix-loop-helix transcription factors used in the article |
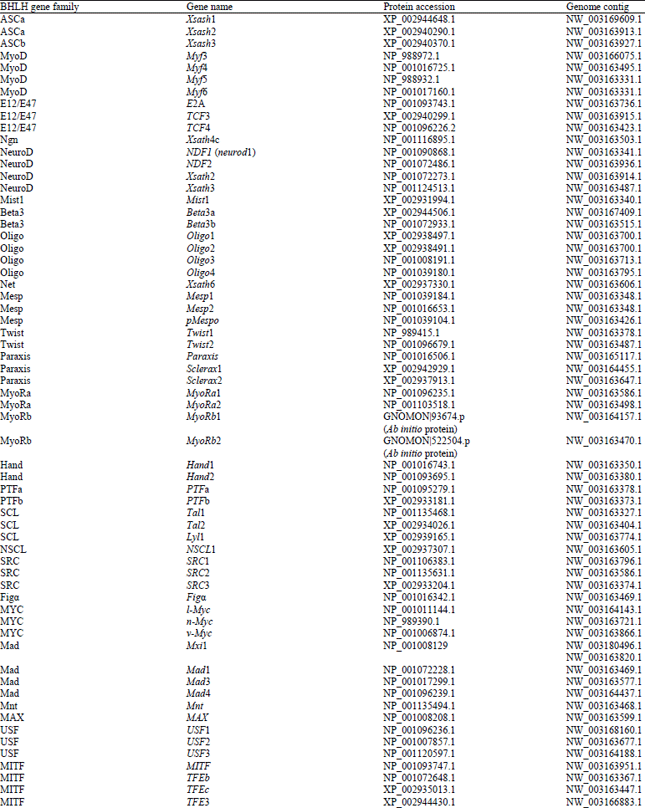  | |
Then, similar BLAST searches were conducted against the Xenbase for putative bHLH proteins too. All of the TBLASTN, BLASTP and PSI-BLAST searches were conducted with the methods and similar parameter setting-ups in the previous works (Liu and Zhao, 2010; Liu, 2011; Liu and Chen, 2013). Subsequently, redundant sequences were removed accordingly.
EST searches: In order to find putative bHLH gene and/or existing Expressed Sequence Tags (ESTs) matching the obtained Xenopus laevis HLH sequences, TBLASTN searches were performed against Xenopus Genome EST database on NCBI and Xenbase TBLASTN websites using each bHLH as the query sequence. The stringency was set as E<0.0001. A 90% or higher identity was considered to be an EST corresponding to the bHLH sequence. The obtained EST was translated into protein sequence with the EditSeq program in DNAStar version 7.1 to obtain the absent amino acid residues. In case where a query sequence composed of two or three Xenopus laevis coding regions, intron splice sites were assessed with the online program NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) (Hebsgaard et al., 1996) to find the locations of possible introns.
Protein ID, sequence alignment and motif comparison: Protein sequence ID (accession number) was obtained by BLASTP searches against the Xenopus laevis protein database with the amino acid sequence of each identified bHLH domain. All of the gained sequences were finally aligned using ClustalX 2.0 (Thompson et al., 1997). The aligned bHLH domains were shaded using GeneDoc 2.6.02 (Nicholas and Nicholas, 1997) and copied into a RTF file for further annotation. Sequences were compared according to conserved amino acids.
Gene Ontology (GO) enrichment analysis: The Gene Ontology (GO) hierarchy annotations were downloaded from the Gene Ontology database (http://www.geneontology.org/). Enrichment for GO term categories was analyzed using DAVID Functional Annotation Bioinformatics Tools (Dennis et al., 2003; Huang et al., 2008) which reports enrichment by scores with respect to GO term categories. DAVID calculates the functional enrichment score of the same gene set based on the GO categories including biological processes, molecular functions, cellular components, KEGG pathways and other key words. In addition, it also provides a hyper-geometric p-value and a Benjamini p-value for each enrichment score.
Phylogenetic analyses of bHLH orthologous genes: Phylogenetic analyses were conducted by MRBAYES 3.1.2 (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) and PHYML 2.4.4 (Guindon and Gascuel, 2003), with the JTT substitution frequency matrix (Jones et al., 1992). The obtained bHLH sequences were used to construct phylogenetic trees of Bayesian inference and maximum likelihood estimation. Maximum Likelihood (ML) analyses were performed using the frequencies of amino acids estimated from the data set and rate heterogeneity across sites modeled by one constant rate and eight γ-rates. Statistical support for the different internal branches was assessed by bootstrap resampling with 100 replicates in PHYML (Guindon and Gascuel, 2003). Phylogenetic analyses of Bayesian inference was performed with two independent Markov chains, each containing from 1200-1500 million Monte Carlo steps until the standard deviation of split frequencies was below 0.01 with sample frequency saved every 1000 generations. Finally, the trees obtained in the two runs of Markov chains were meshed and the first 25% of the trees were discarded as ‘burnin’ and the 50% majority consensus trees were edited and displayed using MEGA 4.0 (Tamura et al., 2007).
RESULTS AND DISCUSSION
Retrieval and identification of bHLH proteins: BLASTP, TBLASTN and PSI-BLAST searches with the 45 representative bHLH domains identified 106 sequences with manual improvement and examination (Table 2, Fig. 1). All of the names and related information of 106 Xenopus laevis bHLH proteins are listed in Table 2, in which six members were identified by EST searches. Each bHLH protein was named according to its phylogenetic relationship with the corresponding human (Homo sapiens) and frog (Xenopus tropicalis) orthologs and paralogs. Where one human or frog bHLH sequence had two or more Xenopus laevis orthologous genes, we used ‘a’, ‘b’ and ‘c’ or ‘1’, ‘2’ and ‘3’ and so on, to number them. For instance, two orthologous genes of the human Hath5 and Hath4a were found in Xenopus laevis. Thus, the Xenopus laevis genes were named Xath5a and Xath5b, Xath4a1 and Xath4a2, accordingly. In this study, it was found that 106 proteins belonged to 39 families with 46, 25, 10, 5, 16 and 3 members in each of the groups A, B, C, D, E and F respectively, while one member belonged to none of these groups and was classified as an ‘orphan’. Members of six families, i.e., Delilah, Mist, Net, MyoRb, PTFa and Trh, were not found in this study. In addition, sixteen hypothetical or predicted proteins were novel bHLH members identified in this study, i.e., NP_001085994.1, NP_001088572.1, NP_001079668.1, NP_001085596.1, NP_001091211.1, NP_001088421.1, NP_001154867.1, NP_001088667.1, NP_001089471.1, NP_001088134.1, NP_001088700.1, NP_001089031.1, NP_001085564.1, NP_001085718.1, NP_001087639.1 and NP_001079757.1. Moreover, three unclearly predicted proteins, i.e. Paraxis (NP_001087941.1), Id3b (NP_001079757.1) and Hes5 (NP_001079464.1) were verified and two misnamed proteins (NP_001079050.1, protein thylacine1, renamed as Mesp2a; NP_001081641.1, protein thylacine2, renamed as Mesp2b) were corrected and re-annotated by TBLASTN searches and robust phylogenetic analyses. Meanwhile, we must caution that our analyses have probably been carried out with unannotated genome assemblies or even ESTs retrieved from the Xenopus Genome Databases built by NCBI and the Xenopus Laevis Genome Project Resources and Xenbase. Therefore, it is possible that we may have missed some bHLHs or have included a few bHLH domains from some pseudogenes. Nevertheless, these data are sufficient for the purpose of this study and alignment of the 106 Xenopus laevis bHLH domains was shown (Fig. 1).
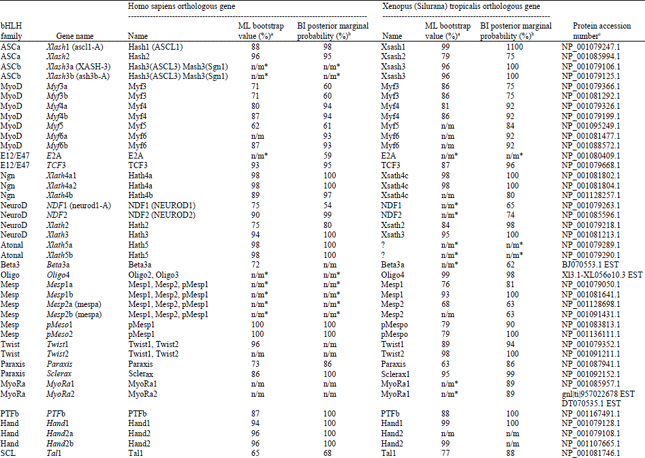
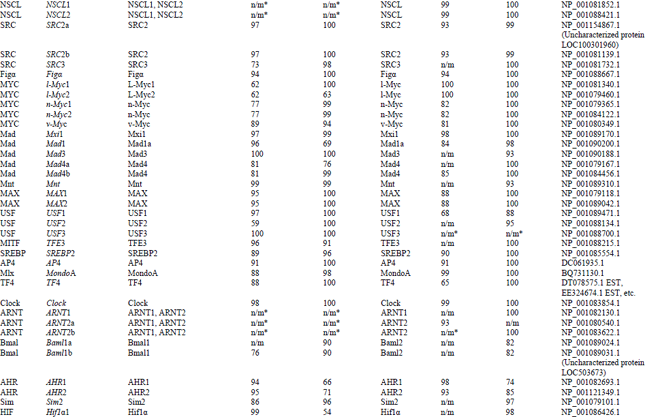
Phylogenetic analyses and identification of orthologous genes: Classification of human bHLH protein members has been extensively studied (Ledent et al., 2002; Simionato et al., 2007; Simionato et al., 2008; Wang et al., 2009; Zheng et al., 2009). Therefore, human bHLH family can be used as a good reference for orthologous gene identification of bHLH members in other organisms. Phylogenetic analysis is still regarded as an effective measure for orthologue identification by constructing phylogenetic trees using robust methods and setting an adequate standard for bootstrap values (Simionato et al., 2007). Determining the phylogenetic relationships of the bHLH proteins is an important step for elucidating the evolutionary and functional divergence of this gene family as well. Herein, phylogenetic analyses of Bayesian Inference (BI) and Maximum Likelihood estimate (ML) were used to identify putative orthologous sequences in different phylogenetic trees with other known bHLH members. If the unknown sequence forms a monophyletic clade with a known bHLH member or family with bootstrap value >50 in phylogenetic trees, the known member will be regarded as orthologous of the putative unknown sequence.
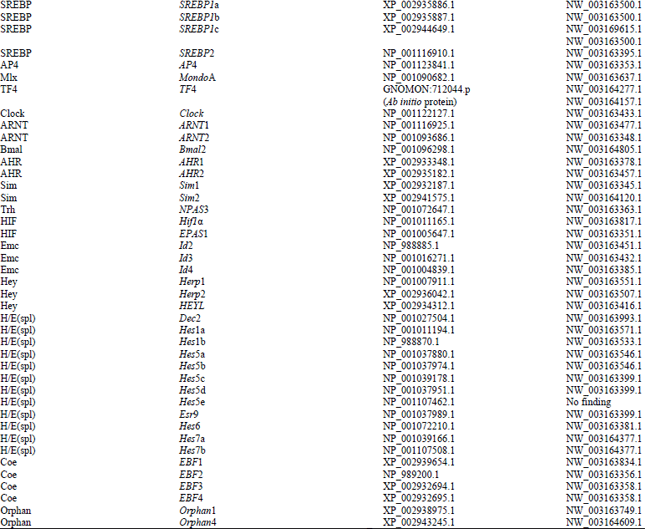
| Table 2: | Information of 106 bHLH genes from Xenopus laevis genome database |
  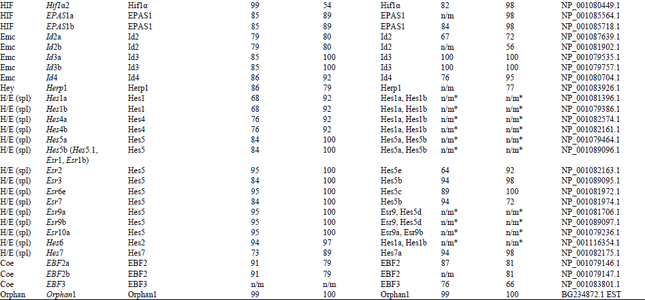 | |
| Xenopus laevis bHLH genes were named according to their human orthologous genes’ names (or common abbreviations) and the reference nomenclature was mainly from the tables and additional tables provided by Ledent et al. (2002). Bootstrap values were inferred and transformed from phylogenetic analyses with human bHLH sequences using Bayesian inference and ML algorithm, respectively. ML bootstrap value (note a) refers the result from maximum likelihood estimate in phylogenetic analysis and BI posterior marginal probability (note b) refers the result from Bayesian inference in phylogenetic analysis. The numbers in the phylogenetic trees are converted into percentages. Note c: The accession numbers were retrieved from two resources. These numbered as ‘NP’ were from the RefSeq protein database and those numbered as ‘XP’ were from the Build protein database. All the bHLH members are organized in the order of bHLH families manifested in Table 2 of Ledent et al. (2002). Notes in the bracket are also gene symbols recorded in NCBI and Xenbase. The question mark means no matching; mark n/m* means none monophyletic group with single particular orthologous gene sequences but formed a monophyletic group with two or more orthologous gene sequences of the family; n/m denotes the case of lower bootstrap value estimated less than 50% | |
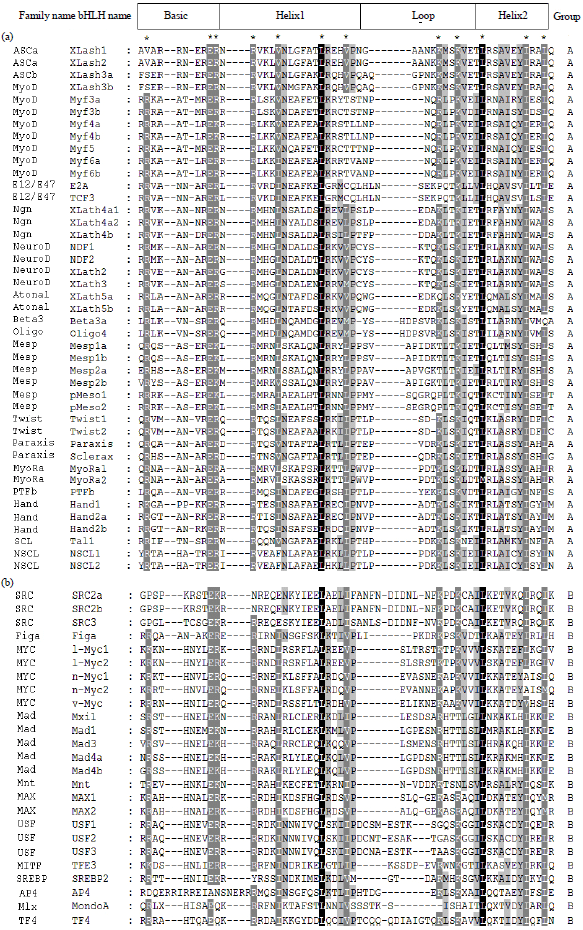  | |
| Fig. 1(a-c): | Alignment of 106 Xenopus laevis bHLH domains shaded using Genedoc, Designation of basic, helix 1, loop and helix 2 follows (Ferre-D'Amare et al., 1993). Family and bHLH protein names and high-order groups were organized according to Table 2 in Ledent et al. (2002). Highly conserved sites are shaded and indicated with asterisks on the top |
This criterion was relaxed for the Mesp, Myc and Hairy/E (spl) families (Morgenstern and Atchley, 1999; Ledent et al., 2002; Simionato et al., 2007, 2008).
In the study, the phylogenetic analyses with 118 human bHLH domains and 105 Xenopus tropicalis bHLH domains revealed that the 106 Xenopus laevis bHLH belong to 39 families in the trees of Bayesian inference and maximum likelihood estimate. The bootstrap values obtained that support the formation of a monophyletic clade with its human orthologous gene are listed in Table 2. The topologies of these BI and ML phylogenetic trees agreed well with each other, though the bootstrap values of maximum likelihood estimate was obtained through 100 replicates. Phylogenetic trees of Maximum Likelihood (ML) estimate and Bayesian inference showed the diversity of the Xenopus laevis bHLH family (Table 2). All the phylogenetic trees are available upon request. It was found that both human and amphibian proteomes have a number of lineage specific or unique bHLH members. For instance, from the Xenopus laevis proteomes, there were no findings of orthologous genes for human Mist, Hath6, MyoRb1 and MyoRb2, PTFa, NPAS3 and Id1 in this study. The Xenopus laevis also have multiple orthologous genes corresponding to one specific human bHLH sequence, such as Hes4a and Hes4b (orthologous genes of human Hes1); Hes5, Esr1a, Esr1b, Esr2, Esr3, Esr6e, Esr7, Esr9a and Esr9b (orthologous genes of human Hes5).
We must caution that our analysis has probably not been carried out on a comprehensive dataset of bHLHs sequences, since the Xenopus laevis lineage experienced a tetraploidization following divergence from the Xenopus tropicalis lineage and the genome project of Xenopus laevis is still under way. In some cases, our search for bHLH genes has been with unannotated genome assemblies or even ESTs retrieved from both the Xenopus Genome Resources built by NCBI and the Xenbase. Therefore, it is possible that we may have missed some bHLHs or that we may have included a few bHLH domains from some duplicates or pseudogenes (Table 2), such as gene duplicates named Myf3a and Myf3b, Myf4a and Myf4b, Myf5, Myf6a and Myf6b, etc. However, these data are sufficient for the purpose of this study. While a few genes showed detectable asymmetric evolution, the majority of genes analyzed in the Xenopus laevis genome were generally consistent with a hypothesis of symmetric change under purifying selection or strong constraint (Hellsten et al., 2007). In addition, there was only a subtle effect affecting on average only about 1-2 amino acids per peptide for any single gene and the relaxed constraint experienced by retained gene duplicates was consistent with their overlapping/redundant biochemical functions (Hellsten et al., 2007).
Analysis of GO enrichment and Functions of bHLH proteins: DNA binding, protein dimerization and transcription coactivator activity are important functional activities of the bHLH domain. In the experiments of site-directed mutagenesis and crystal structures of bHLH proteins, it was shown that Glu-9 and Arg-12 pair constitutes the CANNTG recognition motif. The critical Glu-9 contacts the first CA of the DNA-binding motif and the role of Arg-12 is to fix and stabilize the position of Glu-9 (Fisher and Goding, 1992; Ferre-D'Amare et al., 1993; Ellenberger et al., 1994; Shimizu et al., 1997; Fujii et al., 2000). However, these experiments reported little information of the diverse functions and detailed mechanisms of bHLH proteins and their interactions. To further understand the functions of the Xenopus laevis bHLH gene family systematically, we analyzed the full information and experimental data currently available on the 106 Xenopus laevis bHLH genes by collecting the enrichment records and statistics of Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways with significant hypergeometric p-values and FDR values. Among all the GO terms retrieved, 51 were most significant GO terms (p<0.05) showing key cellular components, molecular functions, biological processes and 5 significant KEGG pathways for the 106 Xenopus laevis bHLH genes (Table 3). Furthermore, each sub-group of the bHLH family has its special gene functions ignoring the common GO term categories (Table 3).
| Table 3: | GO enrichment of each group of Xenopus laevis bHLH genes |
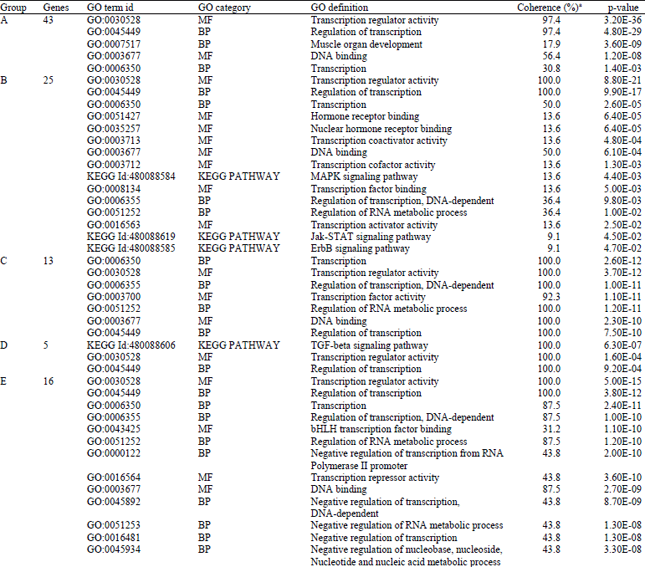 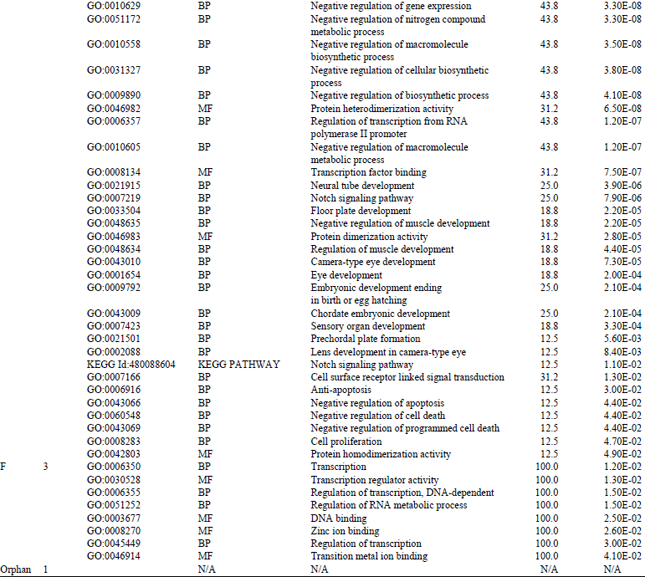 | |
| BP: Biological process; MF: Molecular function. The above table showed the GO annotations enriched significantly (p<0.05, Benjamini corrected Value) in each group. GO annotations included every layer of biological process, molecular function and cellular component category and KEGG pathway. When a GO term and its sub-layer GO both enriched in group significantly, only deeper layer GO annotation is shown in the table. All GO terms in the table were from Gene Ontology database (http://www.geneontology.org). Note a: GO coherence of each group, measured as the percentage of genes in group covered by the GO category | |
Specifically, muscle organ development, neural tube development, chordate embryonic development, embryonic development ending in birth or egg hatching, floor plate development, (negative) regulation of muscle development, nuclear hormone receptor binding, hormone receptor binding, camera-type eye development, eye development, transcription coactivator activity, sensory organ development and Notch signaling pathway also have high frequencies, overlooking the frequent GO categories of transcriptional factors such as regulation of metabolism and biosynthetic processes.
It has been well known that the bHLH genes in various groups have special recognition motifs of DNA-binding sites such as E-box and G-box, etc. So what about the gene functions of each group? To explore this issue, we calculated the hyper-geometric distribution enrichment score of gene molecular functions from group A to group F based on GO annotations of GO categories including the key words of biological process, molecular function, cellular component, KEGG pathways and so on. Only significantly enriched annotations (p<0.05) in deeper layers are shown (Table 3). GO statistics are listed and analyzed with a brief summary of subtypes describing each sub-group too (Table 3). In view of our analysis focused on significant GO terms for the whole frog bHLH gene family and the six bHLH sub-groups (Table 3), we found that each sub-group of bHLH TFs has its own specific GO categories when the common GO terms of transcription such as transcription regulator activity, regulation of transcription and DNA binding and protein dimerization activity are overlooked. Group A is very important in muscle organ development (Table 3). Group B is characterized with hormone receptor binding, nuclear hormone receptor binding, transcription coactivator activity, transcription cofactor activity and a few signaling pathways, i.e., MAPK pathway, Jak-STAT pathway, ErbB pathway and TGF-beta pathway. The members of group C and group D are mainly composed of common GO terms like transcription, transcription regulator activity, transcription factor activity and regulation of transcription, etc. However, group D is evolved in the TGF-beta pathway (Table 3). However, there were only a few GO terms found for members of group C, D and F identified in this study. Group E is composed of some diversified transcription regulators, the GO terms of which are functionally enriched in many aspects of transcription, such as neural tube development, floor plate development, regulation of muscle development, camera-type eye development, eye development, embryonic development ending in birth or egg hatching, chordate embryonic development, sensory organ development, prechordal plate formation, lens development in camera-type eye, cell surface receptor linked signal transduction, anti-apoptosis, cell proliferation, epithelial to mesenchymal transition, neural crest formation, mesenchymal cell development, cell morphogenesis involved in differentiation, identical protein binding, mesenchyme development, mesenchymal cell differentiation, cell morphogenesis, cellular component morphogenesis and Notch signaling pathway. There are also some special GO terms in group F, e.g. DNA binding, zinc ion binding, regulation of transcription, transition metal ion binding, metal ion binding, cation binding and ion binding (Table 3), when omitting the common GO categories of transcriptional factors.
Comparing and analyzing bHLH genes in vertebrate and invertebrate species: With genome sequence data for more and more species becoming available, it is now feasible to compare the bHLH gene family among different animal species at the genomic level. A comparison of bHLH genes in vertebrate and invertebrate species was made across six vertebrate and three invertebrate species (Table 4). Vertebrates have much more bHLH genes than invertebrates and many families in vertebrates have more members, such as E12/E47, NeuroD, Atonal, Mesp, Twist, Paraxis, SCL, SRC, Myc, Mad, MITF, HIF, Emc, Hey and Coe. Among the 45 bHLH families, only 10 families have a single member in human, zebrafish, chicken and rat and mouse respectively, while 33 and 24 families have a single member in lancelet and giant owl limpet (Table 4). It should be noted that, from our result, there are 16 families with one member detected in Xenopus laevis. The Delilah family is missing in vertebrate species and giant owl limpet but exists in Drosophila and Lancelet. It could be attributed to the birth-and-death process of bHLH gene evolution in different vertebrate and invertebrate species (Nei et al., 1997; Nei and Rooney, 2005).
A remarkable group is the H/E (spl) family, including diverse hairy/enhancer of split related genes (mainly Hes proteins and Hes-like factors). In the three invertebrate species, they have either 11 or 12 members, while the vertebrate species have 8-15 members in the H/E (spl) family, respectively. The phylogenetic tree of hairy/enhancer of split like orthologous genes from human, mouse, rat, zebrafish, Xenopus laevis and chicken was explored by a maximum likelihood method with bHLH protein sequences and the zebrafish HEYL being used as out-group (data not shown). It was found that Hes members from human, mouse, rat, zebrafish, Xenopus laevis and chicken form clear monophyletic groups (except human Hes4), indicating that each Hes lineage has its own ancestral sequence. Furthermore, a considerable number of bHLH genes besides the H/E (spl) family were found to have a multi-member distribution pattern in human, mouse, rat, zebrafish, chicken and Xenopus laevis bHLH gene families too. This case suggests that they should arise through gene duplication at least before the divergence of vertebrates from invertebrates. Among those bHLH members, many closely related genes, known as Hes, Her, or ESR and enhancer of split related genes have now been isolated from vertebrates. Like the Drosophila E (spl) genes, many of their vertebrate homologues are expressed in response to Notch activity (Campos-Ortega, 1993, 1994, 1995) and the products of these genes are essential to implement many of the cell fate decisions mediated by Notch signaling, such as the selection of cells to become neural precursors (Artavanis-Tsakonas et al., 1995; Greenwald, 1998). Phylogenetic analysis of the H/E (spl) family revealed that more than four gene duplication events had occurred at an early date. The objective of the next research should be designed to further determine whether accelerated rates of evolution in the H/E (spl) members or bHLH domains are due to increased positive selection or decreased constraint. Although our results provide little support for the positive selection hypothesis, this study provides us an evolutionary scenario in which the diversity of H/E (spl) gene family has been established through possibly relaxed selective constraint (Streisfeld and Rausher, 2007).
| Table 4: | Comparing the number of bHLH transcription factors found among vertebrate and invertebrate species |
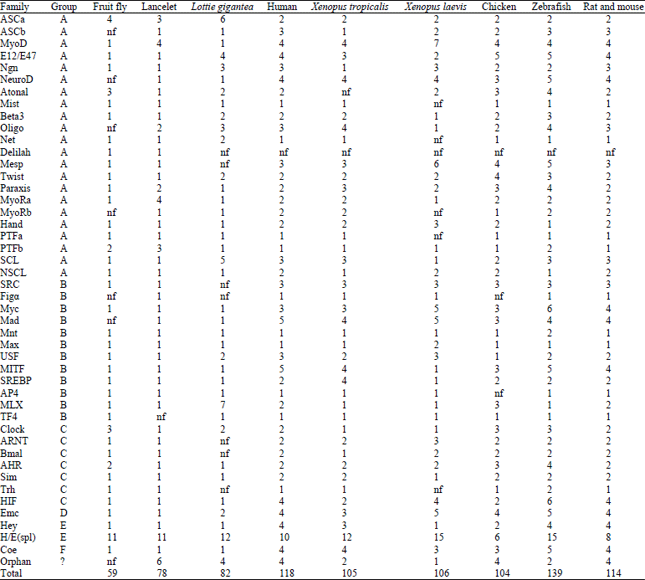 | |
| The vertebrate and invertebrate species referred lancelet (Branchiostoma floridae), Lottie gigantea (Lottie gigantea), fruit fly (Drosophila melanogaster), human (Homo sapiens), zebrafish (Danilo rerio), frog (Xenopus laevis), chicken (Gallus gallus), rat (Rattus norvegicus) and mouse (Mus musculus). Data on lancelet, fruit fly and human are from Simionato et al. (2007). Data on zebrafish, rat and mouse, chicken and Lottie gigantea and Xenopus tropicalis are from Wang et al. (2009), Zheng et al. (2009), Liu and Zhao (2010) and Liu and Chen (2013). Data on Xenopus laevis are from the new data mining in this study. Mark nf denotes the members of particular families were not found in the reported studies | |
CONCLUSION
In the study, we have identified 106 bHLH domains and their protein sequences in the Xenopus laevis genome database by TBLASTN and BLASTP and PSI-BLAST searches with the 45 representative bHLH domains as query sequences. Through phylogenetic analyses of the Xenopus laevis bHLH domains with human bHLH orthologous domains, we assigned the 106 Xenopus laevis bHLH genes into 39 families and an unclassified group termed "orphan". Members of six families, i.e., Delilah, Mist, Net, MyoRb, PTFa and Trh, were not found in the study. Among all the identified bHLH members, these 105 sequences could classified into 39 families with 46, 25, 10, 5, 16 and 3 members in the corresponding high-order groups A, B, C, D, E and F respectively, while one orphan member was found belonging to none of these groups. Furthermore, 16 hypothetical proteins were newly identified and annotated by computational analysis, three unclearly predicted proteins were verified and two misnamed proteins were corrected. Those uncharacterized putative bHLH proteins may be novel transcription factors needing further validation. GO enrichment statistics showed 51 significant GO annotations and 5 significant KEGG categories counted in frequency. In addition, the GO enrichment group-analysis showed that different groups of proteins have their special gene functions when overlooking the common GO term categories.
ACKNOWLEDGMENTS
We are grateful to the anonymous reviewers for their constructive comments and suggestions. It’s jointly funded by the 2014 annual Anhui Provincial Project of Outstanding Young Talents Fund in Colleges and Universities (No.[2014]181), the projects of National Natural Science Foundation of China (No.31071310) and Anhui Provincial Natural Science Foundation (No.1308085QC63) and Anhui Provincial Educational Commission Natural Science Foundation (No. KJ2012A216).
REFERENCES
- Artavanis-Tsakonas, S., K. Matsuno and M.E. Fortini, 1995. Notch signaling. Science, 268: 225-232.
CrossRefPubMedDirect Link - Atchley, W.R. and W.M. Fitch, 1997. A natural classification of the basic helix-loop-helix class of transcription factors. Proc. Natl. Acad. Sci. USA., 94: 5172-5176.
Direct Link - Atchley, W.R., W. Terhalle and A. Dress, 1999. Positional dependence, cliques and predictive motifs in the bHLH protein domain. J. Mol. Evol., 48: 501-516.
CrossRefPubMedDirect Link - Atchley, W.R., K.R. Wollenberg, W.M. Fitch, W. Terhalle and A.W. Dress, 2000. Correlations among amino acid sites in bHLH protein domains: An information theoretic analysis. Mol. Biol. Evol., 17: 164-178.
PubMedDirect Link - Bowes, J.B., K.A. Snyder, E. Segerdell, R. Gibb and C. Jarabek et al., 2008. Xenbase: A Xenopus biology and genomics resource. Nucl. Acids Res., 36: D761-D767.
CrossRefDirect Link - Buck, M.J. and W.R. Atchley, 2003. Phylogenetic analysis of plant basic helix-loop-helix proteins. J. Mol. Evol., 56: 742-750.
CrossRefDirect Link - Campos-Ortega, J.A., 1993. Mechanisms of early neurogenesis in Drosophila melanogaster. J. Neurobiol., 24: 1305-1327.
CrossRefPubMedDirect Link - Campos-Ortega, J.A., 1994. Genetic mechanisms of early neurogenesis in Drosophila melanogaster. J. Physiol. Paris, 88: 111-122.
CrossRefPubMedDirect Link - Campos-Ortega, J.A., 1995. Genetic mechanisms of early neurogenesis in Drosophila melanogaster. Mol. Neurobiol., 10: 75-89.
CrossRefPubMedDirect Link - Carretero-Paulet, L., A. Galstyan, I. Roig-Villanova, J.F. Martinez-Garcia, J.R. Bilbao-Castro and D.L. Robertson, 2010. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in arabidopsis, poplar, rice, moss and Algae. Plant Physiol., 153: 1398-1412.
CrossRefPubMedDirect Link - Carruthers, S. and D.L. Stemple, 2006. Genetic and genomic prospects for Xenopustropicalis research. Seminars Cell Dev. Biol., 17: 146-153.
CrossRefDirect Link - Dennis, G., B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane and R.A. Lempicki, 2003. DAVID: Database for annotation, visualization and integrated discovery. Genome Biol., 4: R60-R60.
CrossRefPubMedDirect Link - Ellenberger, T., D. Fass, M. Arnaud and S.C. Harrison, 1994. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev., 8: 970-980.
CrossRefDirect Link - Ferre-D'Amare, A.R., G.C. Prendergast, E.B. Ziff and S.K. Burley, 1993. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature, 363: 38-45.
CrossRefPubMedDirect Link - Fisher, F. and C.R. Goding, 1992. Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. EMBO J., 11: 4103-4109.
Direct Link - Fujii, Y., T. Shimizu, T. Toda, M. Yanagida and T. Hakoshima, 2000. Structural basis for the diversity of DNA recognition by BZIP transcription factors. Nat. Struct. Biol., 7: 889-893.
CrossRefDirect Link - Greenwald, I., 1998. LIN-12/Notch signaling: Lessons from worms and flies. Genes Dev., 12: 1751-1762.
PubMedDirect Link - Guindon, S. and O. Gascuel, 2003. A simple, fast and accurate algorithm to estimate larges phylogenies by maximum likelihood. Syst. Biol., 52: 696-704.
CrossRef - Hellsten, U., M.K. Khokha, T.C. Grammer, R.M. Harland, P. Richardson and D.S. Rokhsar, 2007. Accelerated gene evolution and subfunctionalization in the pseudotetraploid frog Xenopus laevis. BMC Biol., Vol. 5.
CrossRefDirect Link - Huang, D.W., B.T. Sherman and R.A. Lempicki, 2008. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols, 4: 44-57.
CrossRefPubMedDirect Link - Heim, M.A., M. Jakoby, M. Werber, C. Martin, B. Weisshaar and P.C. Bailey, 2003. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol., 20: 735-747.
CrossRefPubMedDirect Link - Huelsenbeck, J.P. and F. Ronquist, 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17: 754-755.
CrossRefPubMedDirect Link - Jones, D.T., W.R. Taylor and J.M. Thornton, 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci., 3: 275-282.
PubMedDirect Link - Ledent, V. and M. Vervoort, 2001. The basic helix-loop-helix protein family: Comparative genomics and phylogenetic analysis. Genome Res., 5: 754-770.
PubMedDirect Link - Ledent, V., O. Paquet and M. Vervoort, 2002. Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biol.
CrossRefDirect Link - Li, J., Q. Liu, M. Qiu, Y. Pan, Y. Li and T. Shi, 2006. Identification and analysis of the mouse basic/Helix-Loop-Helix transcription factor family. Biochem. Biophys. Res. Commun., 350: 648-656.
CrossRefPubMedDirect Link - Li, X., X. Duan, H. Jiang, Y. Sun and Y. Tang et al., 2006. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and arabidopsis. Plant Physiol., 141: 1167-1184.
CrossRefPubMedDirect Link - Liu, W.Y. and C.J. Zhao, 2010. Genome-wide identification and analysis of the chicken basic helix-loop-helix factors. Comp. Funct. Genomics.
CrossRefDirect Link - Liu, W.Y. and C.J. Zhao, 2011. Molecular phylogenetic analysis of Zebra finch basic Helix-Loop-Helix transcription factors. Biochem. Genet., 49: 226-241.
CrossRefDirect Link - Liu, W.Y., 2011. Genome-wide survey, identification and preliminary analysis of Xenopus Laevis BHLH transcription factors. Hans J. Biomed., 1: 6-16.
CrossRefDirect Link - Liu, A., Y. Wang, C. Dang, D. Zhang, H. Song, Q. Yao and K. Chen, 2012. A genome-wide identification and analysis of the basic helix-loop-helix transcription factors in the ponerine ant, Harpegnathos saltator. BMC Evol. Biol., Vol., 12.
CrossRefDirect Link - Liu, A., Y. Wang, D. Zhang, X. Wang and H. Song et al., 2013. Classification and evolutionary analysis of the basic helix-loop-helix gene family in the green anole lizard, Anolis carolinensis. Mol. Genet. Genomics., 288: 365-380.
CrossRefPubMedDirect Link - Liu, W.Y. and D.Y. Chen, 2013. Phylogeny, functional annotation and protein interaction network analyses of the Xenopus tropicalis basic helix-loop-helix transcription factors. Biomed. Res. Int.
CrossRefDirect Link - Luscombe, N.M., S.E. Austin, H.M. Berman and J.M. Thornton, 2000. An overview of the structures of protein-DNA complexes. Genome Biol., Vol. 1, No. 1.
CrossRefDirect Link - Massari, M.E. and C. Murre, 2000. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol., 20: 429-440.
PubMedDirect Link - Morgenstern, B. and W.R. Atchley, 1999. Evolution of bHLH transcription factors: modular evolution by domain shuffling?. Mol. Biol. Evol., 16: 1654-1663.
PubMedDirect Link - Murre, C., P.S. McCaw and D. Baltimore, 1989. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD and myc proteins. Cell, 56: 777-783.
CrossRefPubMedDirect Link - Nei, M., X. Gu and T. Sitnikova, 1997. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA., 15: 7799-7806.
PubMedDirect Link - Nei, M. and A.P. Rooney, 2005. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet., 39: 121-152.
CrossRefDirect Link - Pires, N. and L. Dolan, 2010. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol., 27: 862-874.
CrossRefPubMedDirect Link - Riechmann, J.L., J. Heard, G. Martin, L. Reuber and C.Z. Jiang et al., 2000. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science, 290: 2105-2110.
CrossRefPubMedDirect Link - Ronquist, F. and J.P. Huelsenbeck, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 12: 1572-1574.
CrossRefPubMedDirect Link - Shimizu, T., A/ Toumoto, K. Ihara, M. Shimizu and Y. Kyogoku et al., 1997. Crystal structure of PHO4 bHLH domain-DNA complex: Flanking base recognition. EMBO J., 16: 4689-4697.
CrossRefDirect Link - Simionato, E., V. Ledent, G. Richards, M. Thomas-Chollier and P. Kerner et al., 2007. Origin and diversification of the basic helix-loop-helix gene family in metazoans: Insights from comparative genomics. BMC Evol. Biol., Vol. 7.
CrossRef - Simionato, E., P. Kerner, N. Dray, M. Le Gouar, V. Ledent, D. Arendt and M. Vervoort, 2008. Atonal-and achaete-scute-related genes in the annelid Platynereis dumerilii: Insights into the evolution of neural basic-Helix-Loop-Helix genes. BMC Evol. Biol.
CrossRef - Stevens, J.D., E.H. Roalson and M.K. Skinner, 2008. Phylogenetic and expression analysis of the basic helix-loop-helix transcription factor gene family: Genomic approach to cellular differentiation. Differentiation, 76: 1006-1022.
CrossRefPubMedDirect Link - Streisfeld, M.A. and M.D. Rausher, 2007. Relaxed constraint and evolutionary rate variation between basic helix-loop-helix floral anthocyanin regulators in Ipomoea. Mol. Biol. Evol., 24: 2816-2826.
CrossRefPubMedDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Toledo-Ortiz, G., E. Huq and P.H. Quail, 2003. The arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell, 15: 1749-1770.
CrossRefPubMedDirect Link - Thompson, J.D., T.J. Gibson, F. Plewniak, F. Jeanmougin and D.G. Higgins, 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25: 4876-4882.
CrossRefPubMedDirect Link - Wang, Y., K. Chen, Q. Yao, W. Wang and Z. Zhu, 2007. The basic helix-loop-helix transcription factor family in Bombyx mori. Dev. Genes Evol., 217: 715-723.
CrossRef - Zheng, X., Y. Wang, Q. Yao, Z. Yang and K. Chen, 2009. A genome-wide survey on basic helix-loop-helix transcription factors in rat and mouse. Mamm. Genome, 20: 236-246.
CrossRefPubMedDirect Link - Bowes, J.B., K.A. Snyder, E. Segerdell, C.J. Jarabek, K. Azam, A.M. Zorn and P.D. Vize, 2010. Xenbase: Gene expression and improved integration. Nucl. Acids Res., 38: D607-D612.
CrossRefDirect Link - Karpinka J.B., J.D. Fortriede, K.A. Burns, C. James-Zorn, V.G. Ponferrada et al., 2015. Xenbase, the Xenopus model organism database; new virtualized system, data types and genomes. Nucl. Acids Res., 43: D756-D763.
CrossRefDirect Link - Hebsgaard, S.M., P.G. Korning, N. Tolstrup, J. Engelbrecht, P. Rouze and S. Brunak, 1996. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res., 24: 3439-3452.
CrossRefPubMedDirect Link