
S. Ahmadi
Department of Physiology, School of Medicine, Kurdistan University of Medical Sciences, Sanandaj, Kurdistan, Iran
L. L. Veinotte
Terry Fox Laboratory, British Columbia Cancer Agency, Canada
Pakistan Journal of Biological Sciences
Year: 2011 | Volume: 14 | Issue: 22 | Page No.: 1002-1010







ABSTRACT
Natural Killer (NK) cells are thought to develop from common lymphoid progenitors in the bone marrow. Even though thymus is not essential for NK cell development, T-cell/natural killer-cell (T/NK) precursors, DN1 (CD44+CD25-) and DN2 (CD44+CD25+) when cultured on an OP9 stroma, give rise to some NK1.1+ cells. Genes of the Schlafen (Slfn) family are involved in hematopoietic and immune processes. The contribution of the Slfn genes in NK cell development from Double Negative (DN) cells is unknown. We transduced DN1 and DN2 progenitors prepared from C57BL/6 (B6) mouse thymus with Schlafen 1 (Slfn1) and Schlafen 2 (Slfn2) genes using Mig retroviral vector containing the Green Fluorescent Protein (GFP) gene and cultured those transduced progenitors on OP9 and OP9 stroma expressing the Notch ligand Delta-like 1 (OP9-DL1) with appropriate cytokines to see if they affect generating NK and T-cells differently. Maturation of both NK and T cells from immature T/NK thymocytes hampered by Slfn1 and Slfn2 transduction but we got a small number of Slfn1 and Slfn2 expressing cells upon culture of transduced DN progenitors on stroma cells. There was no difference between Slfn1 expressing (GFP+) and none expressing T cells regarding CD3 expression but all mature NK cells were from Slfn1 negative population. Slfn2 completely blocked maturation of T cells but there was no difference between Slfn2 expressing and none expressing NK cells. Based on our findings both Slfn1 and Slfn2 interfere with maturation of DN2 progenitors but T cell development is more sensitive to Slfn2 expression than NK cell.
PDF Abstract XML References Citation
Received: August 05, 2011;
Accepted: October 14, 2011;
Published: December 01, 2011
How to cite this article
S. Ahmadi and L. L. Veinotte, 2011. Effect of Schlafen 2 on Natural Killer and T Cell Development from Common T/Natural Killer Progenitors. Pakistan Journal of Biological Sciences, 14: 1002-1010.
DOI: 10.3923/pjbs.2011.1002.1010
URL: https://scialert.net/abstract/?doi=pjbs.2011.1002.1010
DOI: 10.3923/pjbs.2011.1002.1010
URL: https://scialert.net/abstract/?doi=pjbs.2011.1002.1010
INTRODUCTION
Schlafen (Slfn) proteins are paralogous family of proteins with largely unknown function. The mouse Slfn gene family of growth regulatory genes is composed of 10 genes and is thought to arise from a common ancestor through multiple unequal recombination events (Schwarz et al., 1998; Geserick et al., 1998). The function of this newly discovered protein family has been investigated in several studies but so far relatively little is known about the precise functions of Slfn proteins. Genes of the Slfn family are expressed in hematopoietic cells and are involved in regulation of lymphocyte maturation, differentiation, growth control and terminal myeloid differentiation (Schwarz et al., 1998; Geserick et al., 1998; Bruno et al., 2004). Data show that Schlafen1 (Slfn1) and Schlafen2 (Slfn2), are preferentially expressed in the lymphoid lineage and are differentially regulated during thymocyte development. Slfn1 has been reported to play an important role in the establishment and maintenance of quiescence in T-lymphocytes and thymocytes of Slfn1 transgenic mice showed decrease proliferation (Schwarz et al., 1998). Expression of Slfn1 in mouse fibroblasts represses cell growth (Schwarz et al., 1998; Geserick et al., 1998). It was reported that a Slfn1-binding protein named DnaJB6 is necessary for translocation of Slfn1 into the nucleus, where Slfn1 down-regulates cyclin D1 and induces cell-cycle arrest (Zhang et al., 2008). Over expression of cyclin D1 in growth-arrested, Slfn1 expressing cells induced an increase in cell growth consistent with this protein being the biological target of Slfn1 (Brady et al., 2005). Slfn2, the family member bearing the most similarity to Slfn1, was suggested to have a much stronger growth inhibitory activity than Slfn1 (Schwarz et al., 1998).
Generating inducible versions of Slfn2 using the tetracycline-repressible promoter was reported to be unsuccessful (Schwarz et al., 1998). Both Slfn1 and Slfn2 transcript were found to be highly expressing in the thymus, spleen and lymph node but lung and heart (Schwarz et al., 1998). Slfn1 and Slfn2 expression in T cells decreases upon activation with α-CD3 and α-CD28 antibodies. Among differentiating thymocytes, Slfn2 and Slfn1 expression increases during the CD4+CD8+ double positive to single positive transition. Slfn2 has a key functional role in the induction of the growth-suppressive effects of interferons and its knockdown enhances hematopoietic progenitor colony formation and reverses the growth-suppressive effects of interferon alpha on normal hematopoietic progenitors (Katsoulidis et al., 2009). It may play a role in the defense against pathogens through the regulation of quiescence in T cells and monocytes. Mice homozygous for a form of inherited immunodeficiency called Elektra ascribed to Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence (Berger, 2010).
By micro array analysis, we identified only small number of genes differentially expressed in rapidly growing neonatal and adult NK cells cultured with IL-2. Our micro array results showed that adult and neonatal NK cells express low levels of Slfn1 and high levels of Slfn2. The implication of the differential expression of Slfn1 and Slfn2 in adult and neonatal NK cells is still unknown. Relatively high expression of Slfn1 and Slfn2 in resting T cells, their down regulation by T cell activation and growth inhibition by ectopic expression suggests that they may negatively regulate T cell growth. On the other hand, Slfn2 expression, particularly in adult NK cells, is quite high. It is unknown whether NK cells and T cells are differentially regulated by Slfn 2 as no study has been done with Slfn2 in NK cells. Our hypothesis was that Slfn2 differentially regulates NK and T cell development from NK/T progenitors in the thymus as T cell development is more sensitive to inhibition by Slfn2 than NK cell development. To test this, we expressed Slfn2 in immature double-negative 2 (CD4¯CD8 ¯CD44+CD25+) thymocytes (DN2) and tested their effects on NK and T cell development. Slfn1 was used as a control as its inhibitory effect on T cell growth has been well described.
MATERIALS AND METHODS
Materials
Mice: C57BL/6 (B6) mice were bred in our animal facility. Adult mice used in this study were 6 to 10 weeks old. Study protocols were approved by the animal care committee of the University of British Columbia.
Antibodies and cell lines
Antibodies: Phycoerythrin (PE)-conjugated isotype control mouse IgG2a antibody; fluorescein isothiocyanate (FITC)-conjugated isotype control hamster IgG1 antibody; PE-conjugated anti- CD3 (145-2C11); APC-labeled anti-mouse NK1.1 (PK136); CD19, CD3 (145-2C11) and CD4; Streptavidin APC, PE and FITC were purchased from BD-Biosciences (Mississauga, ON). Biotin lineage marker mAbs to mouse CD44 (1M7), CD3ε (145-2C11), CD8, NK1.1, CD19 (1D3), Gr-1(RB6-8C5), Ter119, Mac-1(M1/70), B220 (RA3-6B2), 2.4G2 (anti-FcR?), phycoerythrin (PE)-conjugated anti-CD25 (3C7), isotype control mouse IgG2a (G155-178), rat IgG2b (A95-1), rat IgG2a (R35-95) and IL-2Rβ (TM-β1) were purchased from BD-Biosciences (Mississauga, ON). PE-conjugated anti-mouse CD8α (53-6.7) was purchased from Boehringer Mannheim Biochemica. Interleukin 7 receptor α(IL-7Rα), was purchased from BD Biosciences (Mississauga, ON). For all cell staining and sorting, cells were first preincubated with 2.4G2 hybridoma supernatant (100 μL for 4x106 cells) to block Fc-recptors followed by biotin Lineage marker primary mAbs. All incubations were performed on ice for 30 min and stained cells were analyzed on a FACS Caliber (BD Biosciences, Mississauga, Canada) with the Cell Quest Pro software (BD Biosciences). Cell sorting was carried out on a FACS Vantage SE (BD Biosciences).
Cell lines: OP9 cells were obtained from RIKEN (Tokyo, Japan). OP9 cells transduced with Delta-like 1 and green fluorescent protein (OP9-DL1) were kind gift from Dr. J-C Zuniga Pflucker (Toronto, Canada).
Methods
Microarray sample preparation and analysis: Total RNA was isolated by using the RNeasy Mini Kit (QIAGEN, Mississauga, ON). Double-stranded cDNA was synthesized from total RNA with the Superscript double-stranded cDNA kit (Invitrogen, Carlsbad, CA). The Enzo BioArray high-yield RNA transcript labeling kit (Affymetrix, Santa Clara, CA) produced biotin-labeled cRNA which was fragmented and hybridized to Affymetrix GeneChip Mouse Genome U74Av2 arrays. Microarray experiments were performed at the DNA Array Laboratory, Wine Research Centre, University of British Columbia and at the Affymetrix GeneChip Facility at the Michael Smith Genome Sciences Centre, British Columbia Cancer Agency. All data analysis was performed with Genespring version 7 (Silicon Genetics, Redwood City, CA). Expression values were background corrected, normalized and summarized by using the default settings of the program package (Table 1).
RT-PCR: Total RNA was isolated from the DN1, DN2, Bulk Thymocytes, fibroblast and NK cells with QIAGEN's Rneasy Mini Kit (QIAGEN, Mississauga, ON) and reversed transcribed into cDNA with QIAGEN's Omniscript Reverse Transcription kit.
| Table 1: | Detection of Schlafens gene expression in NK cells by microarray analysis. Gene expression patterns of purified IL-2-activated adult and neonatal NK cells were analyzed in triplicate using Affymetrix GeneChip Mouse Genome U74Av2 arrays |
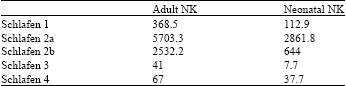 | |
RT-PCR reaction: RNA 10 μL, 10x Buffer RT 2 μL, Oligo dt primer (10 micro molar) 2 μL, RNase inhibitor (10 units/ μL) 2 μL, Omniscript reverse transcriptase 1 μL, RNase- free water 3 μL. The cDNA samples for Reverse Transcriptase (RT)-PCR templates were equal to 100 ng RNA. PCR was done with cDNA using forward primers for Schlafen1 (Slfn1) SlfnORF5 (ATGAACATCACCGATGAAGGG); Schlafen2 (Slfn2); Slfn2B.5 (CTCAGAAAACAGGAGAATGC); Schlafen3 (Slfn3) Slfn3D.5 (ATCAACTCAATCTCAGATGAAG); Schlafen4 (Slfn4) Slfn4.5 (GCAGTTCCTCAAATCCAGAC) and Slfn4/B (GAAGTGAGTGACAGGCAGC) as reverse primer. The reaction volume was 50 μL, containing 5 μL of 10xPCR buffer, 1.5 μL of 50 mM MgCl2, 1 μL of 10 mM dNTPs, 1 μL of each 10 μM forward primers, 3 μL reverse primer, 0.5 μL of 5 U μL-1 Taq DNA polymerase, 36 μL dH2O. Thermocycling condition was as follows: 1 min at 94°C followed by 25 cycles of 30 sec at 94°C, 30 sec at 60°C, 2 min at 72°C and finally 7 min at 72°C. PCR products (10 μL) mixed with 1 μL 10x loading buffer were analyzed on a 1% agarose gel using the Hind III digest of lambda DNA as molecular weight standard (New England Biolabs).
Slfn cDNA Cloning and Plasmid (pBS-Slfn2 and T-Slfn1) Construction
PCR: Slfn2 amplified by polymerase chain reaction using Slfn2B.5, Not1 as forward primers and Slfn4/B, BamH1 as reverse primer. Slfn1 amplified using Sma I and EcoR I containing primers. RT-PCR products (Schlafen genes containing restriction sites) were purified using Wizard PCR preps DNA purification from Promega (Madison, WI). Gel purified Slfn2 DNA and PBS was digested with Not 1 and Bam H1. Gel purified Slfn1 DNA and T vector were digested with Sma I and EcoR I restriction enzymes.
Ligation of Schlafen genes into vectors and Sequencing of PCR products: The PCR products (Slfn2 and Slfn1) were ligated into BamHI and NotI and BamHI and EcoR1 sites of PBS and T vectors respectively.
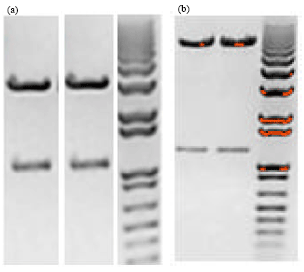 | |
| Fig. 1(a-b): | PCR for cloned plasmids and retroviral constructs. (a): From right, Lane1, 1 kb ladder (λ DNA-HindIII Digest), Lane 2, Double (BamHI and EcoR1) digested T-Slfn1. Two bands as expected. Lane3, Double (BamHI and NotI) digested PBS-Slfn2. Two bands as expected. (b): From right, Lane1, 1 kb ladder (λ DNA-HindIII Digest), Lane 2, Double (BamHI and EcoR1) digested Mig-Slfn1. Two bands as expected. Lane3, Double (BamHI and NotI) digested Mig-Slfn 2. Two bands as expected |
The plasmid clones were sequenced at the NAPS Sequencing Service (University of British Columbia, Vancouver, BC, Canada) and verified using basic local alignment search tool (BLAST). The sequence of clones revealed that clones were true.
Retroviral vector construction (Mig2-Slfn1-IRES-GFP, Mig1-Slfn2-IRES-GFP): Slfn2 cDNA excised from the pBS-Slfn2 plasmid and Slfn1 cDNA excised from Slfn1 T-vector were sub cloned into the Not I, BamHI and BamHI and EcoRI (New England Bio labs) multicloning sites of Mig retroviral expression vector followed by an internal ribosome entry site (Fig. 1).
Isolation of murine NK/T progenitor cells: Single-cell suspensions were prepared from thymuses of young (6-10 weeks old) mice by smashing and passing through a 70 μm filter. Red blood cells were lysed with ammonium chloride solution, cells were washed with 2% PBS and Fc receptors were blocked with 2.4G2 (100 μL for 4x106 cells) hybridoma supernatant (15 min). Thymus cells were depleted of cell surface marker expressing cells. For this purpose thymocytes were stained with biotinylated anti-CD3 and anti-CD8, CD19, Gr-1, Ter119, Mac-1, B220 antibodies for 30 min at 4°C and washed in Phosphate buffer saline. Staining with streptavidin-FITC was performed in the same way.
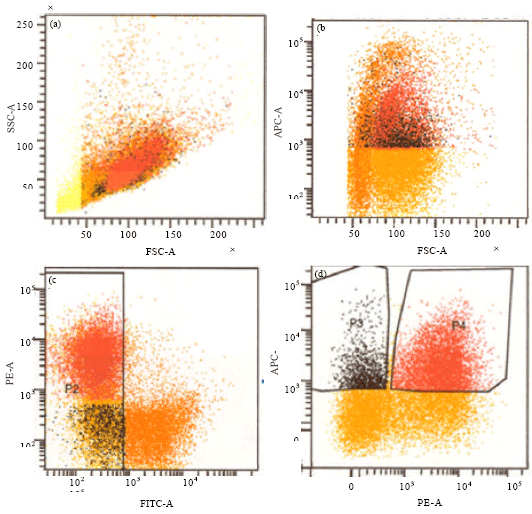 | |
| Fig. 2(a-d): | DN1 (CD44+CD25- ) and DN2 (CD44+CD25+ ) thymocytes isolation by flow cytometry. Lin- cells (i.e. the FITC negative cells) were gated. Out of these DN1 (APC+, PE-) and DN2 (APC+, PE+) cells (P3 and P4 populations) were gated by a fluorescence-activated cell sorting. (a) FSC vs SSC, (b) FSC vs APC, (c) FITC vs PE and (d) PE vs APC |
Positive cells were removed by EasySep FITC Positive Selection kit (Stem Cell Technologies, Vancouver, BC, Canada) and remaining cells (pre-enriched lineage negative cells) were stained with CD25PE, CD44bio-SA-APC and finally propidium iodide was added to 5 μg mL-1. Lin- cells , FITC negative, were gated. Out of these: we sorted DN1 (CD44+CD25- ) CD44 bio-SA-APC single positive and DN2 (CD44+CD25+) CD44 bio-SA-APC CD25PE double positive, cells (Fig. 2) by a Fluorescence-Activated Cell Sorting (FACS) Caliber (BD, Mountain View, CA).
Cell culture, transfections and FACS analysis: Transfection of control mig DNA, Mig-Slfn1-IRES-GFP, Mig-Slfn2-IRES-GFP into GP+E86 murine ecotropic packaging cells and transfecting NIH/3T3 and DN1 and DN2 progenitor cells; Retroviral vectors (Mig2 DNA, Mig-Slfn1-IRES-GFP, Mig2-Slfn2-IRES-GFP) were transfected by calcium-phosphate precipitation into the Phoenix-Eco packaging cell line. For this purpose GP+E86 murine ecotropic packaging cells were maintained in Dulbecco's modified Eagle's medium containing high glucose (4.5 g L-1) and 10% calf serum. Then 20 μg of Mig-Slfn2 or Mig-Slfn1 and empty vector (Mig) DNAs were transfected into 5.0x105 GP+E86 cells in a 60 mm polystyrene dish by calcium phosphate precipitation with a transfection kit (Gibco-BRL). GFP+cells were sorted and cultured. Seventy-two hours later, the supernatant was harvested and filtered through a 0.45 μm filter. To determine the efficiency of transfections , 1 mL of filtered transducing supernatant with 8 μg mL-1 polybrene (Sigma) was added to adherent NIH 3T3 fibroblasts cells seeded at a density of 5*104 cells per well in six-well plates 24 h before the experiment. The plates were centrifuged for 2 h at 1200 g at 30°C. The efficiency of transfections was calculated from the percentage of GFP-positive cells against FSC.
| Table 2: | Expression of Slfn1 and Slfn2 in fibroblasts and analysis of cell growth and viability of cells |
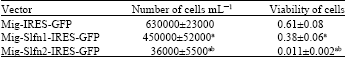 | |
| Expression of Slfn2 led to a significantly reduced recovery in viable cell numbers compared to control and Slfn1 transduced cells. Mig -IRES-GFP stands for empty mig vector, Mig-Slfn1-IRES-GFP stands for Mig vector transduced with Slfn1, Mig-Slfn2-IRES-GFP stands for Mig vector transduced with Slfn2. Data are Mean±SD of experiments repeated three independent times aSignificantly different from empty vector (p≤0.05). bSignificantly different from Slfn1transduced vector (p≤0.05) | |
NIH 3T3 fibroblasts growth analysis: For analysis of the impact of Slfn gene expression on the growth of NIH 3T3 fibroblasts, sub confluent cells were transduced with Mig2 DNA, Mig-Slfn1-IRES-GFP and Mig2-Slfn2-IRES-GFP retroviral vectors. The number of adherent, living cells was determined 3 days after transduction by trypan blue exclusion and the percentage of living cells compared to control transduced cells (Table 2).
Transducing DN2 progenitors: OP9 and OP9-DL1 cells were incubated for 2 days in 6 well plates to perform stroma layers.
For NK cell development, 80000-10000 DN2 cells per well in a 6-well plate seeded with the OP9 were incubated with 500 μL of filtered transducing supernatant from E86 packaging cells expressing retroviral vectors and 500 μL of OP9 media (MEM supplemented with 10% FBS) containing 30 ng mL-1 stem cell factor (SCF), 100 ng mL-1 recombinant human Fms-like tyrosine kinase 3 ligand (Flt3L), 1 ng mL-1 IL-7 and 25 ng mL-1 IL-15 and 5 μg mL-1 polybrene (Sigma) for two hours at 1200 g at 30°C, low acceleration and low brake. For T cell development 80000-10000 DN2 cells per well in a 6-well plate seeded with the OP9-DL1 stroma were incubated with 500 μL of filtered transducing supernatant from E86 packaging cells expressing retroviral vectors and 500 μL of MEM containing 10 ng mL-1 Flt3-L, 5 ng mL-1 IL-7 and 5 μg mL-1 polybrene for two hours at 1200 g at 30°C, low acceleration and low brake. Cells were cultured for 10-14 days before FACS analysis. Half of the medium was removed and changed every 5 days. Harvested cells were incubated with antibodies against Nk 1.1 coupled to APC and CD3 coupled to Phycoerythrin (PE) respectively (Becton-Dickinson). Compensation for GFP, PE and APC was performed with control cells incubated with single antibodies. GFP positive cells were gated and analyzed for CD3 and NK1.1 expression.
RESULTS
Table 1 shows the result of Schlafen genes expression in NK cells by microarray analysis. As it is evident from the results, Slfn2 expression in both neonatal and adult NK cells is high while Slfn1 expression in both neonatal and adult NK cells is low.
Table 2 shows the result of expressing of Slfn1 and Slfn2 in fibroblasts and their effects on cell growth and viability of the fibroblast cells. Expression of Slfn2 led to a significantly reduced recovery in viable cell numbers compared to control and Slfn1 transduced cells.
The result of cut off by a double digestion by restriction enzymes of T-Slfn1 (BamHI and EcoR1), PBS-Slfn2 (BamHI and NotI ), Mig-Slfn1 (BamHI and EcoR1) and Mig-Slfn2 (BamHI and NotI) are shown in Fig. 1a and b, respectively. Analytical gel is shown in Fig. 1 as an evidence of successful cloning.
Figure 2 shows the FACS DN2 progenitors isolated from Lineage negative thymocytes. As it is shown among Lineage negative cells (FITC- cells) CD44+CD25+ (DN2) cells were gated (gate p 2)for transfecting by mig (Fig. 3), Mig-Slfn1 (Fig. 4) and Mig-Slfn2 (Fig. 5) vectors.
Figure 3 shows DN progenitor's transfected with mig vector and cultured on Op9-DL1 or Op9 stroma with appropriate cytokines. The result shows that culture on OP9-DL1 resulted in mature T cells expressing CD3 and culture on the Op9 resulted in mature NK cells expressing NK1.1
Figure 4 shows the result of culturing slfn1 transduced progenitors cultured on OP9-DL1 and OP9 stroma. Population analysis of GFP positive population showed that slfn1 expression blocked maturation of progenitors to mature NK cells expressing NK1.1 (Fig. 4h) but did not affect maturation of DN2 progenitors to mature T cells expressing CD3.
Figure 5 shows the result of expressing Slfn2 in DN progenitors. As it is evident Slfn2 expression prevented maturation of the DN cells to mature T cells (Fig. 5d) but did not affect maturation of DN progenitors to mature NK cells (Fig. 5h).
DISCUSSION
Schlafens are a group of genes involved in the control of cell cycle progression and growth inhibitory responses. They are associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence (Bustos et al., 2009; Gubser et al., 2007). Slfn1 has been identified as a gene which is highly up regulated in positively selected, mature CD4 and CD8 thymocytes as compared to immature progenitors. T cell receptor triggered activation markedly decreases the expression of Slfn1. DN1 and DN2 cells comprise a mixed population of precursors that retains the capacity to differentiate into T and Natural Killer (NK) cells (Balciunaite et al., 2005; Lehar et al., 2005; Porritt et al., 2004; Allman et al., 2003; Pear et al., 2004).
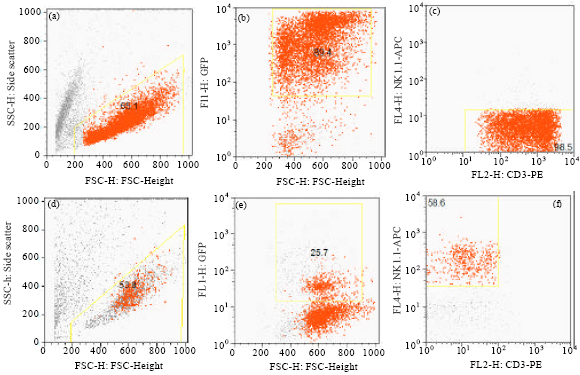 | |
| Fig. 3(a-f): | DN2 cells transduced with mig retroviral vector, cultured on OP9-DL1 (a,b,c) and OP9 (d,e,f) for 14 days in the presence of appropriate cytokines and stained with CD3-PE & NK1.1-APC. a and d (FSC vs SSC), b and e (FSC vs GFP), c and f (CD3-PE vs NK1.1-APC for GFP+ cells) |
Result of our transfection studies of DN2 progenitors with Slfn1 and Slfn2 showed that both Slfn1 and Slfn2 have profound inhibitory effects on the maturation of DN2 progenitors to T cell or NK cells. However, in spite of severe block of progenitor maturation by Slfn1 and Slfn2 expression, small number of Slfn1 and even smaller number of Slfn2 infected DN2 cells finally relieved of growth inhibition and became mature cells. DN2 cells that conquered Slfn1 repression became mature T cell but not NK cells and DN2 cells that conquered Slfn2 repression became mature NK cells but not T cells. Based on the result of this study T cell maturation from DN2 progenitors is more sensitive to Slfn2 expression while NK cell maturation from DN2 progenitors is more sensitive to Slfn1 expression.
It was reported that transgenic Slfn1 expression perturbs thymocyte development, evoking a severe block at the CD25+CD44– stage (DN3) within the immature DN progenitor population (Geserick et al., 1998). Homozygous Slfn1 transgenic mice exhibited a profound loss of single positive thymocytes and T cells. CD4 single positive cells of Slfn1 transgenic mice thymuses are immature and expressed little or no CD3 (Schwarz et al., 1998). Schwarz et al. (1998) reported that they were not able to obtain stable S2-6 transfectants with Slfn2 or Slfn3 which probably indicates that basal expression of Slfn2 or slfn3 is sufficient to completely disrupt cell growth. A reduction in the expression of Slfn1 and Slfn2 following induction of T cell proliferation and the reduction in thymic cellularity in CD2. Slfn1 transgenic mice shows the role of Slfn molecules in the regulation of the cell cycle (Geserick et al., 1998).
Our microarray analysis showed that level of Slfn2 expression in NK cells is higher than T cells. From the culture of DN2 cells transfected with Slfn1 on OP9 stroma with appropriate cytokines we did not get mature NK cells and mature NK cells expressing NK1.1 were all from GFP negative population indicating that Slfn1 transfection of DN2 progenitors completely prevented the maturation of T/NK progenitors to NK cells. Analysis of mature T cells expressing CD3 obtained from the culture of Slfn1 transduced progenitors on OP9-DL1 showed that both GFP positive and GFP negative population express CD3 indicating that some of the Slfn1 transduced T/NK DN progenitors conquered blockage induced by slfn1 expression and became mature T cells. We got mature NK cells from the culture of Slfn2 transfected DN2 progenitors on OP9 stroma but we did not get mature T cells from the culture of Slfn2 transfected DN2 progenitors on OP9-DL1 stroma cells.
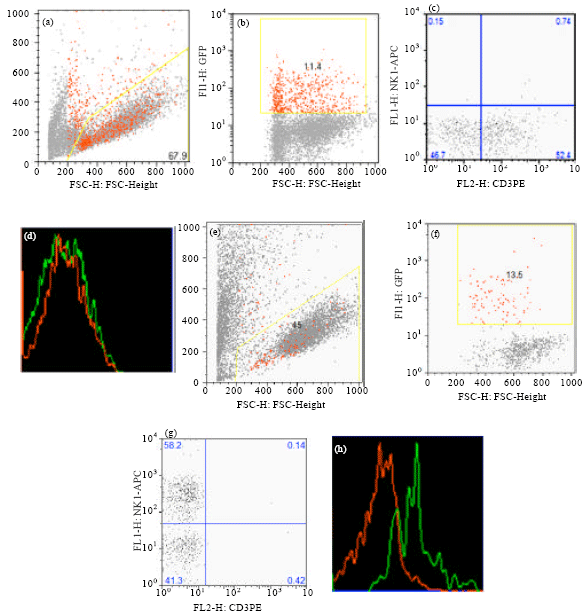 | |
| Fig. 4(a-h): | DN2 cells transduced with Slfn1 and cultured on OP9-DL1 (a-d) and OP9 (e-g) stroma for 14 days in the presence of appropriate cytokines and stained with CD3-PE & NK1.1-APC. (a and e): FSC vs. SSC, (b and f): FSC VS GFP, (c and g): quadrant analysis for CD3-PE and NK1.1-APC, (d): Population analysis for CD3 Expression indicates that CD3 expressing T cells come from both GFP+ (Slfn1 expressing) and GFP- populations. (h): Population analysis for NK1.1 Expression indicates that natural killer cells expressing NK1.1+ come from Slfn1 negative (GFP-) population. Red colors show GFP+ cells and green colors shows GFP- cells |
We got some mature NK cells from Slfn2 expressing DN2 progenitors cultured on OP9 stroma which indicates that even though Slfn2 expressions in general interferes with maturation of DN progenitors, T cell maturation is more sensitive to Slfn2 expression than NK cell maturation.
There have already been some discrepancies about the reported anti-proliferative activities of Slfn genes. It was reported that Slfn1 expressing cells do not undergo apoptosis and the growth inhibition is gradually relieved after four days (Schwarz et al., 1998). Geserick et al. (1998) suggested that Slfn1 expressing cells died eventually of apoptosis. Liang Zhao et al reported that Slfn1 and Slfn2 do not cause any growth inhibition when over expressed in myeloid cells (Zhao et al., 2008). They reported that Slfn1 and Slfn2 do not confer growth inhibition in myeloid cells and NIH/3T3 cells transfected transiently with Slfn1 or Slfn2 expression plasmids showed no G1 cell cycle arrest and arguing that Slfn1 and Slfn2 are unlikely to inhibit Cyclin D1 at transcriptional level.
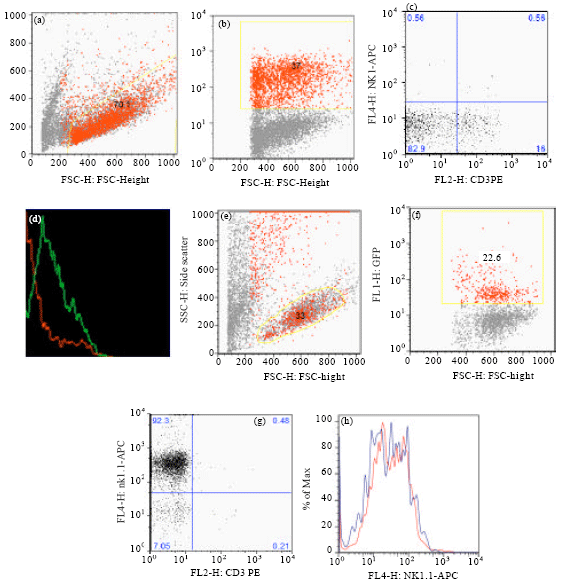 | |
| Fig. 5(a-h): | DN2 cells transduced with Slfn2 and cultured on OP9-DL1 (a-d) and OP9 (e-h) stroma for 14 days in the presence of appropriate cytokines and stained with CD3-PE & NK1.1-APC. (a and e): FSC vs SSC, (b and f): FSC VS GFP, (c and g): quadrant analysis for CD3-PE and NK1.1-APC, (d) Population analysis for CD3 Expression indicates that majority of CD3 expressing T cells come from Slfn2 negative (GFP-) populations. (h): Population analysis for NK1.1 expression indicated that mature NK cells came from both Slfn2 expressing (GFP+) and Slfn2 non expressing cells |
Geserick et al. (1998) reported that they were not able to establish cell lines expressing even minute’s amount of Slfn1 constantly. Liang et al reported that Slfn1 and Slfn2 did not affect cell proliferation regardless of whether over expression was constitutive, induced or from transient transfection. Although early ectopic expression of Slfn1 has a profound effect on thymocyte development, the most prominent attribute of this gene family is their effect on cell growth and progression through the cell cycle. To assess more directly whether Slfn proteins influence cell growth, we expressed Slfn1 and Slfn2 in NIH 3T3 fibroblasts. Our data indicated that the cell growth was less than 10% with Slfn2 and viability of NIH/3T3 infected with Slfn2 was less than 2.5% of those infected with Slfn1 (Table 2). We detected no change in cell viability during Slfn1 expression, as assessed by trypan blue uptake in comparison to empty vector. This appears to exclude apoptosis as the method by which NIH/3T3 growth is disrupted and in agree with what previously reported by others (Schwarz et al., 1998; Geserick et al., 1998). Slfn1 seems to be a negative regulator of cell growth, because its natural or ectopic expression correlates with resting cellular states. Slfn2 effect on cell growth and development is much stronger based on what we observed in our NIH/3T3 cells study. Indeed we reproduced the reported growth inhibition effects of Slfn1 and Slfn2 in NIH/3T3 cells that was reported before in other studies (Schwarz et al., 1998; Geserick et al., 1998) which is different from what reported by Zhao et al. (2008) that believe growth inhibition by Slfn1 and Slfn2 is not reproducible in vitro. The only difference between the result of our work and Geserick et al. (1998) was effect of Slfn1 and Slfn2 on maturation of DN cells to T or NK cells in which T cell maturation from DN2 progenitors was more sensitive to Slfn2 expression while NK cell maturation from DN2 progenitors was more sensitive to Slfn1 expression. Although we still do not fully understand the exact functions of Slfn proteins, our findings emphasize previous notions of regulatory role of Slfn1 and Slfn2 members of this protein family in lymphoid cells. Different response that we got from Slfn1 and Slfn2 regarding NK and T cells maturation from double negative progenitors may contribute to high level of Slfn2 expression in both neonatal and adult NK cells that we saw in our microarray analysis. The regulatory networks guiding both thymic development and differentiation of T cells are complex and the roles of Slfn genes in the processes have only been poorly characterized to date. More studies are needed to better understand the obvious contribution of Slfn proteins in the process of development and differentiation of T cell and NK cells from progenitors.
ACKNOWLEDGMENTS
I would like to thank for Dr. Fumio Takei from Terry Fox Laboratory, British Columbia Cancer Research Center and Department of Pathology and Laboratory Medicine, University of British Columbia for letting me to do this research in his lab. Corresponding author would like to thank of the administrative office in research deputy of Kurdistan University of Medical Sciences for granting sabbatical leave for doing this research in the terry Fox laboratory in University of British Columbia.
REFERENCES
- Schwarz, D.A., C.D.Katayama and S.M. Hedrick, 1998. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity, 9: 657-668.
PubMedDirect Link - Bruno, L., R. Hoffmann, F. McBlane, J. Brown and R. Gupta et al., 2004. Molecular signatures of self-renewal, differentiation, and lineage choice in multipotential hemopoietic progenitor cells in vitro. Mol. Cell. Biol., 24: 741-756.
PubMedDirect Link - Zhang, Y., Z. Yang, Y. Cao, S. Zhang and H. Li et al., 2008. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem. J., 413: 239-250.
PubMed - Brady, G., L. Boggan, A. Bowie and L.A. O'Neill, 2005. Schlafen-1 causes a cell cycle arrest by inhibiting induction of cyclin D1. J. Biol. Chem., 280: 30723-30734.
PubMed - Katsoulidis, E., N. Carayol, J. Woodard, I. Konieczna and B. Majchrzak-Kita et al., 2009. Role of Schlafen 2 (SLFN2) in the generation of interferon α-induced growth inhibitory responses. J. Biol. Chem., 284: 25051-25064.
PubMed - Berger, M., P. Krebs, K. Crozat, X. Li and B.A. Croker et al., 2010. An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nature Immunol., 11: 335-343.
CrossRefDirect Link - Bustos, O., S. Naik, G. Ayers and C. Casola and M.A. Perez-Lamigueiro et al., 2009. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene, 447: 1-11.
PubMed - Gubser, C., R. Goodbody, A. Ecker, G. Brady, L.A.J. O'Neill, N. Jacobs and G.L. Smith, 2007. Camelpox virus encodes a schlafen-like protein that affects orthopoxvirus virulence. J. Gen. Virol., 88: 1667-1676.
Direct Link - Zhao, L., B. Neumann, K. Murphy, J. Silke and T.J. Gonda, 2008. Lack of reproducible growth inhibition by Schlafen1 and Schlafen2 in vitro. Blood Cells Mol. Dis., 41: 188-193.
PubMedDirect Link - Balciunaite, G., R. Ceredig and A.G. Rolink, 2005. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage and natural killer cell but no B-lymphocyte potential. Blood, 105: 1930-1936.
Direct Link - Lehar, S.M., J. Dooley, A.G. Farr and M.J. Bevan, 2005. Notch ligands Delta1 and Jagged1 transmit distinct signals to T-cell precursors. Blood, 105: 1440-1447.
PubMed - Porritt, H.E., L.L. Rumfelt, S. Tabrizifard, T.M. Schmitt, J.C. Zuniga-Pflucker and H.T. Petrie, 2004. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity, 20: 735-745.
PubMed - Allman, D., A. Sambandam, S. Kim, J.P. Miller and A. Pagan et al., 2003. Thymopoiesis independent of common lymphoid progenitors. Nat. Immunol., 4: 168-174.
PubMed - Pear, W.S., L. Tu and P.L. Stein, 2004. Lineage choices in the developing thymus: Choosing the T and NKT pathways. Curr. Opin. Immunol., 16: 167-173.
PubMed - Geserick, P., F. Kaiser, U. Klemm, S.H. Kaufmann and J. Zerrahn, 1998. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int. Immunol., 16: 1535-1548.
PubMedDirect Link