
M. Islami
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
A. Shabani
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
M. Saifi-Abolhassan
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
Sh. Sepehr
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
M.R. Soudi
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
S.Z. Mossavi-Nejad
Department of Biology, Faculty of Basic Science, Alzahra University, Tehran, Iran
Pakistan Journal of Biological Sciences
Year: 2008 | Volume: 11 | Issue: 2 | Page No.: 208-213







ABSTRACT
Purification and characterization of alcohol dehydrogenase (ADH) from Gluconobacter suboxydans was done in order to biotechnological and industrial application. Solubilization of enzyme from bacterial membrane fraction by Triton X-100 and subsequent fractionation on DEAE-Sephadex A-50 and Hydroxyapatite was successful in enzyme purification. Enzyme assay reaction mixture contained potassium ferricyanide 0.1 M, McIlvaine buffer 0.1 M (pH 5.5), Triton X-100 10%, ethanol 1 M and enzyme solution. The purified ADH Optimum pH activity was 5.5. The enzyme was in maximum stability in pH 5.8. The substrate specificity of the enzyme was determined using the same enzyme assay method as described above, except that various substrates (100 mM) were used instead of ethanol. The relative activity of the ADH for ethanol was higher than the others. The effects of metal ions and inhibitors on the activity of the enzyme were examined by measuring the activity using the same assay method as described above. Activity of purified enzyme was increased in the presence of Ca+2 and was decreased in presence the of ethylenediamine tetra acetic acid (EDTA). Because the proper structure and function of the enzyme is related to structural Ca+2 and EDTA can chelate Ca+2. An apparent Michaelis constant for ethanol were examined to be 1.7x10-3 M for ethanol as substrate.
PDF Abstract XML References Citation
How to cite this article
M. Islami, A. Shabani, M. Saifi-Abolhassan, Sh. Sepehr, M.R. Soudi and S.Z. Mossavi-Nejad, 2008. Purification and Characterization of Alcohol Dehydrogenase from Gluconobacter suboxydans. Pakistan Journal of Biological Sciences, 11: 208-213.
DOI: 10.3923/pjbs.2008.208.213
URL: https://scialert.net/abstract/?doi=pjbs.2008.208.213
DOI: 10.3923/pjbs.2008.208.213
URL: https://scialert.net/abstract/?doi=pjbs.2008.208.213
INTRODUCTION
Quinoproteins are oxidoreductases that posses one of the four different o-quinone cofactor family, including pyrroloquinoline quinine (PQQ), tryptophan tryptophylquinone (TTQ), trihydroxyphenylalanyl quinone (topaquinone or TPQ), lysine tyrosylquinone (LTQ) and cysteine tryptophylquinone (CTQ) instead of nicotinamide or flavine cofactors (Salisbury et al., 1979; Duine, 1991; Ameyama et al., 1981; Adachi et al., 2003; Anthony, 1992; Cai et al., 1997).
In many prokaryotic organisms, various simple sugar and alcohol dehydrogenases have noncovalent PQQ cofactor (Davidson, 1993; Duine et al., 1990; Goodwin and Anthony, 1998). The enzyme quinoproteins, have certain properties which make them superior to other dehydrogenases in vinegar fermentation, 2-keto-L- gulconic acid and 5-ketogluconic acid production (both of which can be easily converted to vitamin C) (Mutsushita et al., 2002; Saeki et al., 1997) and analytical applications, especially in biosensor applications (D`Costa et al., 1986).
There are two major types of PQQ-containing alcohol dehydrogenases (ADHs) (EC 1.1.99.) with and without heme group. Methanol dehydrogenase (MDH) in methylotrophs and type I alcohol dehydrogenase (ADH I) in Pseudomonas species have only PQQ as cofactor (Adachi et al., 1998). In the case of quinohemoprotein ADH, some is present as a free-form of a single protein called type II ADH (ADH II), while the other, called type III ADH(ADH III) is as a complex with a cytochrome c subunit. Type II ADH has been found in Pseudomonas and related species, while type III ADH only in acetic acid bacteria including the genera Actobacter and Gluconobacter (Adachi et al., 1987a, b; Mutsushita et al., 2002). ADH III is a quinohemoprotein-cytochrom c complex bound to the periplasmic side of the cytoplasmic membrane and function as the primary dehydrogenase in ethanol oxidase respiratory chain, where ADH oxidizes ethanol by transferring electrons to ubiquinon embedded in the membrane phospholipids (Matsushita et al., 1992; Ameyama et al., 1981).
Coupled with ethanol oxidation, ADH reduces phenazine methosulfate, dichlorophenolindophenol, or ferricyanide as an artificial electron acceptor in vitro (Ameyama and Adachi, 1982). Since ferricyanide reacts with the heme components having a high redox potential, the heme c sites in ADH complex should reduce ferricyanide (Matsushita et al., 1992).
In this study, a successful example of a complete purification of a quihemoprotein membrane-bound ethanol dehydrogenase has been described for Iranian Gluconobacter suboxydans. In addition, some properties of the purified membrane-bound ethanol dehydrogenase (EtDH) have been presented.
MATERIALS AND METHODS
Chemicals: All chemicals used in this study were commercial products.
DEAE-Sephadex, Hydroxyapatite and Potassium ferricyanide were purchased from Sigma Chemical Company. Sodium gluconate and Potato extract were kind from microbiology lab of Alzahra University, Total Protein Assay Kit from Chem Enzyme Company.
Microorganism: As the enzyme source of purification of the PQQ alcohol dehydrogenase, the bacterium, Gluconobacter suboxydans was purchased from Persian Type Culture Collection at IROST Iran (PTCC).
Medium and cultivation: Basal medium employed in this study contained 20 g of sodium gluconate, 5 g D-glucose, 3 g of glycerol, 3 g yeast extract, 2 g polypeptone, 200 mL of potato extract in liter of tap water (pH 7). The type culture of acetic acid bacteria grown on the yeast extract slant was inoculated to 100 mL of the medium in 500 mL shaking flask and the cultivation was carried out at 37°C for 24 h with reciprocal shaking.
Enzyme assay: EtDH was assayed using potassium ferricyanide as an electron acceptor. The rate of reduction of ferricyanide to ferrocyanide gives a quantitative amount of ethanol oxidation. The reaction mixture contained 0.1 mL potassium ferricyanide 0.1 M, 0.5 mL McIlvaine buffer 0.1 M, pH 5.5, 0.1 mL Triton X-100 10%, 0.1 mL ethyl alcohol 1 M and enzyme solution in total volume of 1 mL. The reaction started by the addition of ethanol solution at 25°C and stopped by adding 0.5 mL of the ferric-dopanol reagent after 5 min. Then, 3.5 mL of water was further added to the last mixture and well mixed. The resulting stabilized Prussian blue color formed was measured by spectrophotometer at 660 nm after standing for 20 min at 25°C. One unit of enzyme activity was defined as amount of enzyme catalyzing the oxidation of 1 μmol of ethanol per min under these assay conditions and 4.0 absorbance unit equaled to 1 μmol of ethanol oxidized (Adachi et al., 1987a, b).
Protein assay: The protein concentration was estimated by measuring by Total Protein Chem Enzyme Assay Kit.
Protein concentration in sample (g dL-1) = (Sample observation/standard observation)xstandard concentration
SDS-polyacrylamide gel electrophoresis (SDS-PAGE): For estimation of purity of enzyme preparations, slab gel electrophoresis was performed under essentially the same conditions as described by Laemmli (1970) using 12.5% of polyacrylamide gel and Tris HCL buffer, pH 8.3, sodium dodecyl sulfate (SDS)-acrylamide gel electrophoresis was performed to determine purity and subunit composition of the enzyme (Laemmli, 1970).
RESULTS
Preparation of cell homogenate: Cells were harvested by centrifugation at 12,000 x g for 20 min and washed with saline 0.9%. The cell paste was suspended in 0.01 M potassium phosphate buffer, pH 6.0, (1 g of wet cell/10 mL buffer) and this suspension was sonicated with sonicator at 100 W for 5 steps (5 min) with intervolves (2 min). Intact cells was removed by centrifugation at 5000 x g for 5 min. The resulting supernatant was disintegrated as cell homogenate.
Solubilization of enzyme: The membrane fraction is suspended in 0.01 M buffer, pH 6.0 and the protein concentration is adjusted to 30 mg mL-1 Triton X-100 and 2-mercaptoethanol are added to final concentrations of 1.0% and 1 mM, respectively. The suspension is gently stirred for 3 h at 0°C and centrifuged at 68,000 x g for 60 min. Supernatant is obtained as the solubilized enzyme.
DEAE-sephadex column chromatography (I): To the solubilized enzyme solution, polyethylene glycol 6000 is added to 20% to precipitate the enzyme. After 30 min of stirring in an ice bath, the enzyme solution is centrifuged at 12,000 x g for 20 min. The precipitate is suspended in small volume of 0.01 M buffer and the thick suspension is dialyzed overnight against 0.002 M buffer containing 0.1% Triton X-100. The dialyzed solution is applied to a DEAE-Sephadex A50 column (5x30) that has been equilibrated with 0.002 M buffer, pH 6.0, containing 0.1% Triton X-100. The column is washed with 500 mL of the same buffer to remove nonadsorbable materials. The enzyme is eluted from the column with 0.1 M buffer, pH 6.0, containing 1% Triton X-100. Pooled enzyme fraction in dialyzing tubing is concentrated by dehydration by embedding the enzyme in dry polyethylene glycol 6000. The concentrated faction is then dialyzed thoroughly against 0.002 M buffer, pH 6.0, containing 0.1% Triton X-100. The insoluble material is removed by centrifugation at 12,000 g for 20 min. Chromatography on DEAE-Sephadex A-50(II) was repeated (Fig. 1).
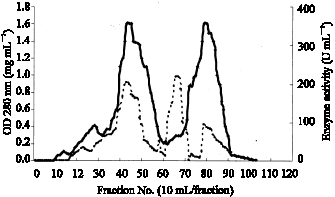 | |
Fig. 1: | Chromatography on DEAE-Sephadex A50 (I) ADH solution from proceeding step was adsorbed onto column of DEAE-Sephadex A50 (I) (5x30) that has been equilibrated with 0.002M buffer, pH 6.0, containing 0.1% Triton X-100. The enzyme is eluted from the column with 0.1 M buffer, pH 6.0, containing 1% Triton X-100. (___) was enzyme activity and ( - - - ) was protein content |
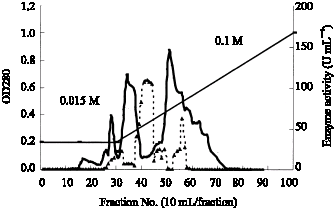 | |
Fig. 2: | Chromatography on DEAE-Sephadex A50 (II) ADH solution from DEAE-Sephadex A50 (I) was adsorbed onto column of DEAE-Sephadex A50 (II) (1.5x20), which has been equilibrated with the 0.015M buffer, pH 6.0, containing 0.05% Triton X-100 and elution of the enzyme was performed by a linear gradient elution made between 0.015M and 0.1M of phosphate buffer. (___) was enzyme activity and ( - - -) was protein content |
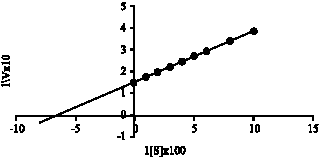 | |
Fig. 3: | Lineweaver-Burk graph of oxidation of ethanol by EtDH. Enzyme activity was measured at various concentration of ethanol as indicated |
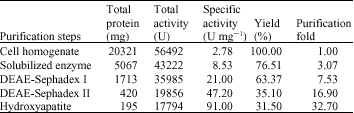
Table 1: | A summary of the purification steps of the enzymes |
 | |
eae-sephadex column chromatography (II): The dialyzed enzyme is applied to the second column DEAE-Sephadex A50 (1.5x20), which has been equilibrated with the 0.015 M buffer, pH 6.0, containing 0.05% Triton X-100 and elution of the enzyme was performed by a linear gradient elution made between 0.015 and 0.1 M of phosphate buffer. Each buffer reservoir contained 500 mL and 0.05% Triton X-100, pH 6.0, was present throughout this step. This second step of DEAE-Sephadex A-50 chromatography was found convenient to bring about in removing an impurity which was fractionated in the next step. Pooled enzyme fraction was dialyzed against 0.01 M buffer containing 0.05% Triton X-100 overnight (Fig. 2).
Hydroxyapatite fractionation: The dialyzed enzyme from preceding step was applied to a fractionation with Hydroxyapatite, which had been equilibrated with 0.01 M buffer, pH 6.0, containing 0.05% Triton X-100. We can pour the solution on the Hydroxyapatite, after gently mixing, the mixture was stayed to 5 h for adsorption the enzyme to the beads of Hydroxyapatite. Elution of the enzyme was made stepwise with 0.02, 0.05 and 0.1 M buffer, pH 6.0, containing 0.1% Triton X-100. Pooled enzyme solution was dialyzed 0.002 M buffer thoroughly (2 days). In Table 1, the steps of purification is summarized.
Kinetic analysis of enzyme activity: A steady-state kinetic analysis of the ADH reaction was performed in 100 mM KPB (pH 6.0). To determine the apparent Km value for ethyl alcohol, its concentration was varied from 10 to 100 μM. An apparent Michaelis constant for ethanol were examined to be 1.7x10-3 M (Fig. 3).
Substrate specificity: The substrate specificity of the enzyme was determined using the same enzyme assay method as described above, except that various substrate solutions (100 mM) include methanol, ethanol, isopropanol, n-butanol, formaldehyde, benzaldehyde, glycerol, D-glucose, D-fructose, lactate. The data have been shown in Table 2.
Effects of metal ions and EDTA: The effects of metal ions and inhibitors on the activity of the enzyme were examined by measuring the activity using the same assay method as described above. Each compound solution was
Table 2: | Substrate specificity of the purified enzyme from Gluconobacter suboxydans. The reaction rate with ethanol is expressed as 100 |
 | |
Table 3: | Effect of EDTA and metals on the activity of the purified enzyme. The reaction rate without any additive is expressed as 100 |
 | |
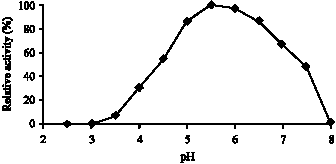 | |
Fig. 4: | Optimum pH of EtDH. Enzyme activity was assayed under standard conditions except that pH of the buffer (MacIlvain buffer) was varied as indicated above. The enzyme shows maximum activity at pH 5.5 |
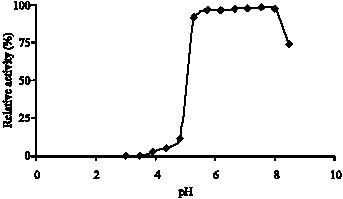 | |
Fig. 5: | Stability pH graph of EtDH. Enzyme solution was diluted with various pH of MacIlvaine buffer from 2.5 to 8 as indicated and stored for 24 h at 4°C. Thereafter, an aliquot of stored enzyme solution was picked up for the standard assay of enzyme activity performed at pH 5.5. EtDH was in maximum stability in pH 5-8 |
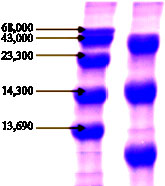 |
| Fig. 6: | SDS-gel electrophoresis of ADH. 20 microliter of the last step purified enzyme solution was loaded on the top of the gel (right). Standard marker solution (left) was contained bovine serum albumin, ovalbumine, trypsine, lysoyzme and ribonuclease A from top to bottom, respectively |
tirred into the reaction mixture and the reaction was started with the addition of the enzyme. Each compound was added to the reaction mixture at a concentration of 1.0 mM, except that the concentration of EDTA was 5.0 mM. The data have been shown in Table 3.
Optimal pH and pH stability: The correlation between the reaction rate of the ADH and pH values of the reaction mixture was determined by the same assay method as described above, except that various pHs and buffers were used.
The enzyme optimal pH graph was showed in Fig. 4 and enzyme pH stability graph was showed in Fig. 5.
Electrophoretic analysis: Dissociation into subunits was observed by SDS gel electrophoresis in the determination of molecular weight of the enzyme. In the present of SDS the enzyme was dissociated into three subunits with a molecular weight 44, 14.3 and 12.5 kD from the top to bottom of the gel column as shown in Fig. 6. The sum of molecular weight of each band gave 70.8 kD of total molecular weight.
DISCUSSION
Alcohol Dehydrogenase (ADH) of acetic acid bacteria, consisting of the genera Actobacter and Gluconobacter catalyzes the first step of acetic acid production, oxidation of ethanol to acetaldehyde (Duine and Frank, 1981; Olsthoorn and Duine, 1996; Salisbury et al., 1979).
This study was an attempt to purify and characterize membrane bound EtDH from Iranian Gluconobacter suboxydans. In PQQ ADH, PQQ bound noncovalent but tightly to the enzyme, whereas in NAD-dependent ADH, NAD serves as a noncovalent cofactor that loosely bound to the enzyme. So, PQQ ADH is more suitable and cheaper than NAD-dependent ADH for industrial and biotechnological applications.
Purified enzyme has been shown to possess substrate specificity for primary aliphatic alcohol. Primary aliphatic alcohol was rapidly oxidized but not methanol. Ethanol is best substrate for ADH. With concern the tertiary structure of methanol dehydrogenase (MDH) and (EtDH), the volume of active site cavity, where substrate bind and react on the top of MDH, EtDH, yielding 18 and 62 A°, respectively. These numbers are well correlated with the substrate specificity. MDH have rather narrow substrate specificity, while EtDH react well with a relatively larger alcohol as well as ethanol. In addition above, the substrate would probably enter through the hydrophobic mouth of a channel leading to the active site cavity and located between PQQ and heme-domains in the case of EtDH. In the case of EtDH, one amino acid residue that helps to form a hydrophobic wall for the active site cavity is located in the heme-domain. Thus, we can say methanol can not properly enter the hydrophobic mouth of a channel leading to the active site cavity of EtDH (Hirohida et al., 2004).
So, the enzyme is suitable for acetic acid industrial production. Secondary and tertiary alcohols and cyclic alcohol could not oxidize.
ADH activity has been shown to increase in the presence of 1 mM Ca+2 and decrease in presence the same concentration of ethylenediamine tetra acetic acid (EDTA). Experiments clearly indicate that calcium is required for catalysis in PQQ containing enzymes. In agreement with a catalytic role, calcium does not seem to be involved in the binding of the substrate. Instead, may polarize the PQQ C5-O5 bond together with the active site Arg residue, resulting in a partial negative charge on the O5 atom and a partial positive charge on the C5 atom (Anthony, 1996) Because the proper structure and function of the enzyme is related to structural Ca+2 and EDTA can chelate Ca+2. The enzyme has a pH optimum at 5-6.5 and enzyme activity was decreased at pH<5 and pH>6.5. That`s reasons may be substrate or enzyme or both not suitable ionic form or enzyme inactivation or all of them. The enzyme stability pH was at pH 5-8.5. Therefore, pH 5-6.5 may be used for the enzyme activity control.
REFERENCES
- Baricevic, D., S. Sosa, R.D. Loggia, A. Tubaro, B. Simonovska, A. Krasna and A. Zupancic, 2001. Topical anti-inflammatory activity of Salvia officinalis L. leaves: The relevance of ursolic acid. J. Ethnopharmacol., 75: 125-132.
Direct Link - Glamoclija, J., M. Sokovic, J. Vukojevic, I. Milenkovic and L.J.L.D. van Griensven, 2006. Chemical composition and antifungal activities of essential oils of Satureja thymbra L. and Salvia pomifera sp. calycina (Sm.). Hayek J. Essential Oil Res., 18: 115-117.
Direct Link - Hammer, K.A., C.F. Carson and T.V. Riley, 1999. Antimicrobial activity of essential oils and other plant extracts. J. Applied Microbiol., 86: 985-990.
CrossRefPubMedDirect Link - Hohmann, J., I. Zupko, D. Redei, M. Csanyi, G. Falkay, I. Mathe and G. Janicsak, 1999. Protective effects of the aerial parts of Salvia officinalis, Melissa officinalis and Lavandula angustifolia and their constituents against enzyme-dependent and enzyme-independent lipid peroxidation. Plant. Med., 65: 576-578.
Direct Link - Horiuchi, K., S. Shiota, T. Hatano, T. Yoshida, T. Kuroda and T. Tsuchiya, 2007. Antimicrobial activity of oleanolic acid from Salvia officinalis and related compounds on vancomycin-resistant enterococci (VRE). Biol. Pharm. Bull., 30: 1147-1149.
Direct Link - Imanshahidi, M. and H. Hosseinzadeh, 2006. The pharmacological effects of Salvia species on the central nervous system. Phytother. Res., 20: 427-437.
Direct Link - Koga, T., N. Hirota and K. Takumi, 1999. Bactericidal activities of essential oils of basil and sage against a range of bacteria and the effect of these essential oils on Vibrio parahaemolyticus. Microbiol. Res., 154: 267-273.
CrossRefDirect Link - Miladinovi , D., 2000. Antimicrobial activity of essential oil of sage from Serbia. Facta. Univ. Ser: Phys. Chem. Technol., 2: 97-100.
Direct Link - O'Mahony, R., H. Al-Khtheeri, D. Weerasekera, N. Fernando, D. Vaira, J. Holton and C. Basset, 2005. Bactericidal and anti-adhesive properties of culinary and medicinal plants against Helicobacter pylori. World. J. Gastroenterol., 11: 7499-7507.
Direct Link - Pereira, R.S., T.C. Sumita, M.R. Furlan, A.O. Jorge and M. Ueno, 2004. Antibacterial activity of essential oils on microorganisms isolated from urinary tract infection. Rev. Saude Publ., 38: 326-328.
CrossRefPubMedDirect Link - Sivropoulou, A., C. Nikolaou, E. Papanikolaou, S. Kokkini, T. Lanaras and M. Arsenakis, 1997. Antimicrobial, cytotoxic and antiviral activities of Salvia fructicosa essential oil. J. Agric. Food Chem., 45: 3197-3201.
CrossRefDirect Link - Shirazi, M.H., R. Ranjbar, S. Eshraghi, G. Sadeghi, N. Jonaidi, N. Bazzaz and N. Sadeghifard, 2007. An evaluation of antibacterial activity of glycyrrhiza glabra Extract on the growth of Salmonella, Shigella and ETEC E. coli. J. Biol. Sci., 7: 827-829.
CrossRefDirect Link - Valero, M. and M.C. Salmeron, 2003. Antibacterial activity of 11 essential oils against Bacillus cereus in tyndallized carrot broth. Int. J. Food Microbiol., 85: 73-81.
CrossRefPubMedDirect Link