
Mohammad Dilmaghani Zadeh
Department of Biochemistry, Faculty of Medicine, Iran University of Medical Sciences, P.O. Box 14155-5983, Tehran, Iran
Reza Amini
Department of Biochemistry, Faculty of Medicine, Iran University of Medical Sciences, P.O. Box 14155-5983, Tehran, Iran
Mohsen Firoozray
Department of Biochemistry, Faculty of Medicine, Iran University of Medical Sciences, P.O. Box 14155-5983, Tehran, Iran
Pupak Derakhshandeh-Peykar
Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Sciences, P.O. Box 14155-1595, Tehran, Iran
Pakistan Journal of Biological Sciences
Year: 2007 | Volume: 10 | Issue: 23 | Page No.: 4246-4250







ABSTRACT
Homozygous deletion is the main mechanism of CDKN2A gene inactivation in malignant gliomas. However different frequencies were reported for its deletion. In order to find the homozygous deletion frequency among Iranian patients, we have analyzed the status of CDKN2A gene in 40 malignant gliomas and examined their 1α and 2 exons by comparative multiplex Polymerase Chain Reaction (PCR), using D9S171 chromosomal marker as an internal control. We found homozygous deletion in 6 out of 7 cases (85.7%) of anaplastic astrocytomas and 20 out of 33 cases (60.6%) of glioblastoma multiforme, in total 26 out of 40 cases (65%) of malignant gliomas. We also found that CDKN2A deleted patients were younger than CDKN2A non-deleted patients and that exon 2 was deleted more than exon 1α.
PDF Abstract XML References Citation
How to cite this article
Mohammad Dilmaghani Zadeh, Reza Amini, Mohsen Firoozray and Pupak Derakhshandeh-Peykar, 2007. Frequent Homozygous Deletion of p16/CDKN2A Gene in Malignant Gliomas of Iranian Patients. Pakistan Journal of Biological Sciences, 10: 4246-4250.
DOI: 10.3923/pjbs.2007.4246.4250
URL: https://scialert.net/abstract/?doi=pjbs.2007.4246.4250
DOI: 10.3923/pjbs.2007.4246.4250
URL: https://scialert.net/abstract/?doi=pjbs.2007.4246.4250
INTRODUCTION
Gliomas are the most common brain tumors (Maher et al., 2001). Astrocytoma types of gliomas were classified by WHO into three grades in order of their malignancy: Grade II or astrocytoma, grade III or Anaplastic Astrocytoma (AA) and the most malignant grade IV or Glioblastoma Multiforme (GBM) (Louis et al., 2007). Progression from low-grade glioma (grade II) to malignant gliomas (grades III and IV) is associated with CDKN2A gene alterations which is inactivated mainly via homozygous deletion (Nakamura et al., 2001), although hypermethylation (Rocco and Sidransky, 2001) and rare mutations (Ichimura et al., 2000) are alternate mechanisms of its inactivation.
It has been shown that the survival rate of patients with malignant gliomas was 40% at one year (Ohgaki et al., 2004). Their poor prognosis makes them good targets for new therapeutic methods like gene therapy. In a study by Wang et al. (2001), p16INK4A expressing vectors has improved survival in animal models of glioma, even when compared with p53 expressing vectors. The frequency of CDKN2A homozygous deletion could be a guide for future gene therapy projects using p16INK4A expressing vectors in malignant gliomas.
Homozygous deletion of CDKN2A gene was not found in germline of familial gliomas (Tachibana et al., 2000), although germline point mutations or microdeletions of CDKN2A gene were reported in about 20% of familial malignant melanoma (Goldstein, 2004). In addition, except for the proven causes of brain tumors which are rare hereditary syndromes and therapeutic radiation, no other significant risk factor has been described for gliomas (Wrensch et al., 2002).
The CDKN2A gene encodes a tumor suppressor known as p16INK4A that binds to CDK4 and CDK6 and inhibits cyclin D attachment to them, which normally leads to pRb hypophosphorylation and cell cycle arrest (Vermeulen et al., 2003). Inactivation of p16INK4A allows cells to escape cell cycle arrest in G1, thus it seems that it could not be inactivated in normal cells including normal glial cells.
The CDKN2A gene is 27.5 kb in length, it is located on 9p21 chromosomal band and consists of three exons, 1α, 2 and 3. It generates three transcripts, two of which encode similar proteins and the third transcript with an alternate first exon (1α), encodes for a different protein known as p14ARF. Although homozygous deletion is the main mechanism of CDKN2A gene inactivation in malignant gliomas, however different frequencies of homozygous deletion have been reported. The highest frequency (82%) has been reported by Barker et al. (1997) and the lowest frequency (15%) by Mochizuki et al. (1999).
In this study, we examined the status of CDKN2A gene exons 1α and 2 of 40 Iranian patients by comparative multiplex PCR and found that they are frequently deleted in malignant gliomas of Iranian patients.
MATERIALS AND METHODS
The study was done in department of Medical Genetics of Tehran Medical Sciences University. Tumor samples were collected from patients with no history of familial glioma who undergone tumor resection surgery from 2003 till 2006, in six different hospitals from Tehran. All specimens were formalin-fixed, paraffin-embedded and graded according to the WHO classification. The analyzed samples consisted of 7 anaplastic astrocytomas and 33 glioblastoma multiformes. DNA samples were isolated by deparaffination followed by proteinase K digestion and phenolchloroform extraction as described previously (Shi et al., 2002).
Comparative multiplex polymerase chain reaction was used to assess the homozygous deletion of exons 1α and 2 of CDKN2A gene. The D9S171 microsatellite marker was selected as the internal control because of its proximity to the target gene, which provides a similar exposure rate for both the target and the control to the DNA polymerase and because it was never found to be homozygously deleted (Xing et al., 1999). The sequences of oligonucleotide primers (Isogen Life Sciences) were: 5'-GAGCAGCATGGAGCCTTC-3' (sense) and 5'-AATTCCCCTGCAAACTTCGT-3' (antisense) for exon 1α and 5'-CACTCTCACCCGACCCGT-3' (sense) and 5'-ACCTTCCGCGGCATCTAT-3' (antisense) for exon 2. Primers used for the amplification of internal control D9S171 were: 5'-AGCTAAGTGAACCTCATCTCTGTCT-3' (sense) and 5'-ACCCTAGCACTGATGGTATAGTCT-3' (antisense). The PCR amplification was performed in 25 μL reaction volumes with about 200 ng of template DNA, 1 μM of each primer, 0.02 μ μLG1 Taq DNA polymerase (Cinnagen), 200 μM of each deoxynucleotide triphosphate, 1.5 mM MgCl2, 50 mM Tris-HCl (pH 8.3), 250 mM KCl and 5% dimethyl sulfoxide. All PCR reactions were done in a programmable thermal cycler (Eppendorf Master Cycler 5330) with the following conditions: initial denaturation at 94°C for 5 min, followed by amplification cycles consisting of denaturation at 94°C for 45 sec, annealing at 54°C for 45 sec, extension at 72°C for 45 sec and final extension at 72°C for 5 min. The cycle number for amplification of exon 1α and D9S171 were 31 and for exon 2 and D9S171 were 32. PCR products were run on a 2% agarose gel, stained with 0.5 μg mLG1 ethidium bromide and visualized under ultraviolet illumination. The intensity of the bands was compared using ImageQuant TL software (Amersham Biosciences). All results were analyzed with SPSS statistical software.
RESULTS AND DISCUSSION
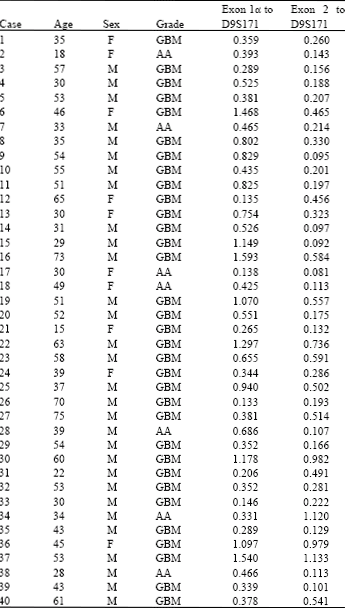
Statistical analysis of the age of the patients with glioma grade, by t-test revealed a possible correlation between them (p = 0.018). The mean age of patients with anaplastic astrocytoma and glioblastoma multiforme were 33.0"9.6 and 47.5"14.9 years, respectively (Table 1).
| Table 1: | Summary of the patients characteristics and PCR product intensity ratios |
 | |
| Table 2: | Spectrum of deletions, according to tumor grades and the sex of the patients |
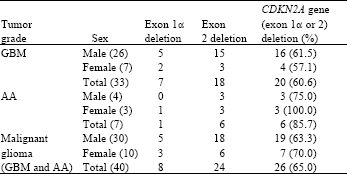 | |
Based upon similar studies (Xing et al., 1999; Kamiryo et al., 2002; Kraus et al., 2000) 0.30 was defined as an arbitrary cutoff for determining the deletion of the target gene in examined samples. Patients with PCR products intensity ratios below this threshold were considered to have lost their CDKN2A gene. Thus, of the 40 malignant gliomas, exon 1α was deleted in 8 cases (20%), 7 of which were GBM and 1 was AA. Exon 2 was deleted in 24 cases (60%), 18 of which were GBM and 6 were AA. Finally, 26 cases (65%) showed deletion in one of the exons 1α or 2, 20 of which were GBM and 6 were AA (Table 2). The incidence of CDKN2A gene deletion for females and males were 70% (7 out of 10) and 63.3% (19 out of 30), respectively. Fisher's exact test showed no correlation between sex and CDKN2A deletion (p>0.05). In contrast a correlation between CDKN2A gene deletion and patient age was found by t-test examination (p = 0.043). The mean ages of the CDKN2A gene deleted and non-deleted patients were 41.5"14.5 and 51.5"14.4 years, respectively (Fig. 3).
Examples of the multiplex PCRs for exon 1α and 2 (Fig. 1, 2). In Fig. 1, exon 1α is deleted in lanes 3 and 4 which correspond to tumor samples 17 and 21 with bands intensity ratios of 0.138 and 0.265, respectively (Fig. 1). Exon 2 is deleted in lanes 2, 3, 4 and 5 which are correspondent with tumor samples 24, 39, 17 and 21 with bands intensity ratios of 0.286, 0.101, 0.081 and 0.132, respectively (Fig. 2).
GBMs were clinically divided into two entities. Primary or de novo GBMs present with no prior history and affect mainly the elderly and secondary GBMs which progress from gliomas of lower grades over a number of years and manifest in younger patients. Although histologically indistinguishable, two distinct genetic pathways are involved in their genesis. Primary GBMs are genetically characterized by LOH 10q, EGFR amplification, CDKN2A homozygous deletion and PTEN mutations (Ohgaki, 2005). Secondary GBMs develop through progression from low-grade astrocytoma or AA.
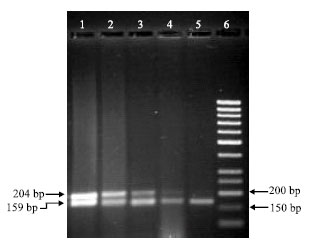 | |
| Fig. 1: | The gel image of exon 1α and D9S171 multiplex PCR. Lane numbers correspond to samples as follow: 1, 31; 2, 11; 3, 4; 4, 21; 5, 17 and 6, molecular weigh marker (Lf 50). Exon 1α is deleted in samples 17 and 21 (Lanes 4 and 5). PCR products were run on a 2% agarose gel (at 100 volt for 45 min) |
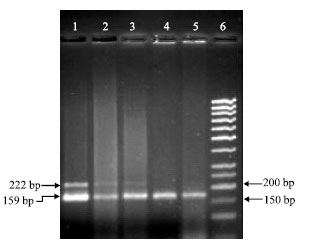 | |
| Fig. 2: | The gel image of exon 2 and D9S171 multiplex PCR. Lane numbers correspond to samples as follow: 1, 19; 2, 24; 3, 39; 4, 17; 5, 21 and 6, molecular weigh marker (Lf 50). Exon 2 is deleted in samples 24, 39, 17 and 21 (Lanes 2, 3, 4 and 5). PCR products were run on a 2% agarose gel (at 100 volt for 45 min) |
In the pathway to secondary GBM, TP53 mutations are the most frequent and earliest detectable genetic alteration, although LOH 10q, CDKN2A, p14 and RB1 inactivation through deletion or methylation are frequently observed (Ohgaki, 2005).
Different studies were done on 9p21 locus which contains this gene in low and high-grade gliomas. Bigner et al. (1988) showed a high incidence of chromosomal 9p structural rearrangement in high-grade gliomas.
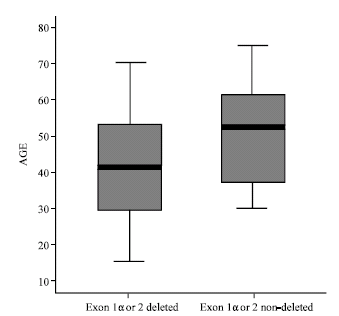 | |
| Fig. 3: | Possible correlation between age and CDKN2A gene deletion (p = 0.043). Patients with exons 1α or 2 deletions were younger (mean age 41.5"14.5 years) than patients without deletions (mean age 51.5"14.4 years) |
Olopade et al. (1992), reported molecular evidence of deletion in 9p in 10 of 15 glioma-derived cell lines and 13 of 35 primary gliomas. Nobori et al. (1994) were the first who reported CDKN2A homozygous deletion in 61.5% of their analyzed glioma specimens. The frequency reported by Barker et al. (1997) was 82% among Caucasians. By contrast, such a frequency was 34.6% in Chinese, reported by Guang and Xianhou (1998) and the lowest frequency (15%) was found among Japanese by Mochizuki et al. (1999).
Here, we report the first study on the CDKN2A gene homozygous deletion frequency in malignant glioma of Iranian patients. In this study, we examined 40 malignant gliomas and we found that 26 of them (65%) had lost their CDKN2A gene exons 1α or 2. We also found that exon 2 was more deleted than exon 1α in both tumor grades (p = 0.000).
We analyzed the band intensity ratios by cutoffs of 0.25 and 0.35 and found the frequency of CDKN2A deletion as 57.5 and 72.5%, respectively. In fact, with both thresholds, CDKN2A gene is frequently lost in malignant gliomas among Iranian patients. Although these frequencies are greater than the frequency reported for the Japanese patients, it is lower than that reported for the Caucasian patients. The obtained results are based on the assumption that the reduced intensity of the CDKN2A band compared to the internal control, corresponds to a reduced amount of template DNA containing the CDKN2A gene.
Analyzing malignant glioma samples we found that patients with CDKN2A gene deletion were younger (p = 0.043). It has been shown that the majority of GBM cases (>90%) are primary glioblastomas that develop rapidly de novo (Ohgaki, 2005) and that CDKN2A gene homozygous deletion occurs in primary GBM more frequently than secondary GBM (Biernat et al., 1997). Although two entities of GBM were not separated in current study, high frequency of the CDKN2A gene homozygous deletion could result from the high number of primary GBMs. Here we suggest that p16INK4A inactivity could be an important mechanism in primary GBM gliomagenesis in young patients.
Present study shows that the rate of exon 2 deletion is higher than exon 1α deletion and a significant difference exists between them, analyzed by the McNemar=s test (p = 0.000). This could be due to an alteration in p14ARF gene which shares exon 2 with CDKN2A gene. Further studies are underway to investigate other mechanisms responsible for p16INK4A inactivity in gliomas that do not have homozygous deletion.
ACKNOWLEDGMENTS
We thank Dr. Alireza Sadeghipoor for helping with specimen collection and classification of tumor grades. We also thank Dr. Mehrdad Pedram (Tehran University of Medical Sciences (TUMS), Tehran, Iran) for his careful proofreading and critical reviews of manuscript.
REFERENCES
- Barker, F.G., P. Chen, F. Furman, K.D. Aldape, M.S. Edwards and M.A. Israel, 1997. p16 deletion and mutation analysis in human brain tumors. J. Neurooncol., 3: 17-23.
CrossRefDirect Link - Biernat, W., Y. Tohma, Y. Yonekawa, P. Kleihues and H. Ohgaki, 1997. Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol., 94: 303-309.
CrossRefDirect Link - Bigner, S.H., J. Mark, P.C. Burger, M.S. Jr. Mahaley, D.E. Bullard and L.H. Muhlbaier, D.D. Bigner, 1988. Specific chromosomal abnormalities in malignant human gliomas. Cancer Res., 48: 405-411.
Direct Link - Goldstein, A.M., 2004. Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum. Mutat., 23: 630-630.
PubMedDirect Link - Zhai, G. and X. Yuan, 1998. Study of deletion of p16 gene in the progression of brain astrocytomas. Chin. J. Cancer Res., 10: 284-287.
CrossRefDirect Link - Ichimura, K., M.B. Bolin, H.M. Goike, E.E. Schmidt, A. Moshref and V.P. Collins, 2000. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res., 60: 417-424.
PubMedDirect Link - Kamiryo, T., K. Tada, S. Shiraishi, N. Shinojima and H. Nakamaura et al., 2002. Analysis of homozygous deletion of the p16 gene and correlation with survival in patients with glioblastoma multiforme. J. Neurosurg., 96: 815-822.
Direct Link - Kraus, J.A., N. Glesmann, M. Beck, D. Krex, T. Klockgether, G. Schackert and U. Schlegel, 2000. Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long-term survivors of glioblastoma multiforme. J. Neurooncol., 48: 89-94.
CrossRefDirect Link - Louis, D.N., H. Ohgaki, O.D. Wiestler, W.K. Cavenee and P.C. Burger et al., 2007. The 2007 WHO classifcation of tumours of the central nervous system. Acta Neuropathol., 114: 97-109.
CrossRefDirect Link - Maher, E.A., F.B. Furnari, R.M. Bachoo, D.H. Rowitch, D.N. Louis and W.K. Cavenee, R.A. DePinho, 2001. Malignant glioma: Genetics and biology of a grave matter. Genes Dev., 15: 1311-1333.
CrossRefPubMedDirect Link - Zadeh, M.D., R. Amini, M. Firoozray and P. Derakhshandeh-Peykar, 2007. Frequent homozygous deletion of p16/CDKN2A gene in malignant gliomas of Iranian patients. Pak. J. Biol. Sci., 10: 4246-4250.
CrossRefDirect Link - Nakamura, M., T. Watanabe, U. Klangby, C. Asker and K. Wiman et al., 2001. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol., 11: 159-168.
CrossRefDirect Link - Nobori, T., K. Miura, D.J. Wu, A. Lois, K. Takabayashi and D.A. Carson, 1994. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature, 368: 753-756.
CrossRefPubMedDirect Link - Ohgaki, H., P. Dessen, B. Jourde, S. Horstmann and T. Nishikawa et al., 2004. Genetic pathways to glioblastoma: A population-based study. Cancer Res., 64: 6892-6899.
Direct Link - Rocco, J.W. and D. Sidransky, 2001. p16(MTS-1/CDKN2A/INK4A) in cancer progression. Exp. Cell Res., 264: 42-55.
CrossRefDirect Link - Shi, S.R., R.J. Cote, L. Wu, C. Liu and R. Datar et al., 2002. DNA extraction from archival formalin-fixed, paraffin-embedded tissue sections based on the antigen retrieval principle: Heating under the influence of pH. J. Histochem. Cytochem., 50: 1005-1011.
Direct Link - Tachibana, I., J.S. Smith, K. Sato, S.M. Hosek, D.W. Kimmel and R.B. Jenkins, 2000. Investigation of germline PTEN, p53, p16INK4Ap14ARF and CDK4 alterations in familial glioma. Am. J. Med. Genet. A, 92: 136-141.
CrossRefDirect Link - Vermeulen, K., D.R. van Bockstaele and Z.N. Berneman, 2003. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif., 36: 131-149.
PubMedDirect Link - Wang, T.J., M.S. Huang, C.Y. Hong, V. Tse, G.D. Silverberg and M. Hsiao, 2001. Comparisons of tumor suppressor p53, p21 and p16 gene therapy effects on glioblastoma tumorigenicity in situ. Biochem. Biophys. Res. Commun., 287: 173-180.
PubMedDirect Link - Wrensch, M., Y. Minn, T. Chew, M. Bondy and M.S. Berger, 2002. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro. Oncol., 4: 278-299.
CrossRefDirect Link