
Fazlul Huq
School of Biomedical Sciences, Faculty of Health Sciences, The University of Sydney, Australia
Journal of Pharmacology and Toxicology
Year: 2006 | Volume: 1 | Issue: 5 | Page No.: 456-463







ABSTRACT
Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that there are both electron-rich and electron-deficient regions on the molecular surfaces of IBF and its metabolites so that they can be subject to both electrophilic and nucleophilic attacks. The latter attack means that they can react with cellular glutathione, thus causing glutathione depletion and hence oxidative stress, and can also cause oxidation of nucleobases in DNA and thus DNA damage. However, the large LUMO-HOMO energy differences observed for IBF and all its metabolites may mean that the rates of such adverse reactions may be low.
PDF Abstract XML References
How to cite this article
Fazlul Huq, 2006. Molecular Modelling Analysis of the Metabolism of Ibuprofen. Journal of Pharmacology and Toxicology, 1: 456-463.
DOI: 10.3923/jpt.2006.456.463
URL: https://scialert.net/abstract/?doi=jpt.2006.456.463
DOI: 10.3923/jpt.2006.456.463
URL: https://scialert.net/abstract/?doi=jpt.2006.456.463
INTRODUCTION
Ibuprofen (IBP, 2-(4-isobutylphenyl)propanoic acid) is a non-steroidal anti-inflammatory drug (NSAID) that is widely used in humans and veterinary medicine in the treatment of acute and chronic rheumatoid arthritis (Rorarius et al., 1993). It is the drug of choice among the ‘profen’ class of NSAIDs because of its low unwanted side effects (Hao et al., 2005). It is also used as a paediatric anti-inflammatory agent (Bennet et al., 1992).
IBP is a chiral molecule that can exist in R- and S-configurations. Metabolic chiral inversion is actually a common characteristic of other arylpropanoic derivatives such as ketoprofen, flubiprofen and pranoprofen (Hao et al. 2005). It is believed this metabolic activation of IBF through chiral inversion not only leads to higher therapeutic potency but may also cause a greater risk of acute kidney failure in patients with renal disorders (Marasco et al., 1987). The adverse effects of IBF include gastrointestinal disturbances and central nervous system depression; however, the severity is found to be mild (McElwee et al., 1990).
The metabolism of IBF in man involves conjugation with glucuronic acid to form ibuprofen acylglucuronide (IBF-GLU) which is stereoselective to S-enantiomer (Lee et al., 1985), oxidation to produce two major metabolites, 2-hydroxyibuprofen (2HIBF) and carboxyibuprofen (CIBF) (Adams et al., 1967; Mills et al., 1973; Brooks and Gilbert 1974; Kaiser et al., 1976; Paterson et al., 1978). The urinary excretion of free IBF, IBF-GLU, 2HIBF and CIBF accounts for between 74 to 86% of an oral dose following the administration of the racemic mixture to man. Three other minor oxidation products: 1-hydroxibuprofen (1HIBF), 3-hydroxyibuprofen (3HIBF) and 2-(4-carboxyphenyl) propanoic acid (CPPA) have also been identified in human urine. The formation of possible metabolite 2-(4-hydroxymethylphenyl) propanoic acid (HPPA) remains speculative as it has not been detected. CYP2C9 is the predominant enzyme responsible for the oxidative metabolism of IBF as it is true for any other NSAIDs (Tracy et al. 1995). Figure 1 shows the metabolic pathway of IBF in humans.
In this study, molecular modelling analyses have been carried out using the program Spartan ’02 (2002) to investigate the relative stability of IBF and its metabolites with the aim of providing a better understanding on their relative toxicity. The study was carried out in the School of Biomedical Sciences, The University of Sydney during January to June 2006.
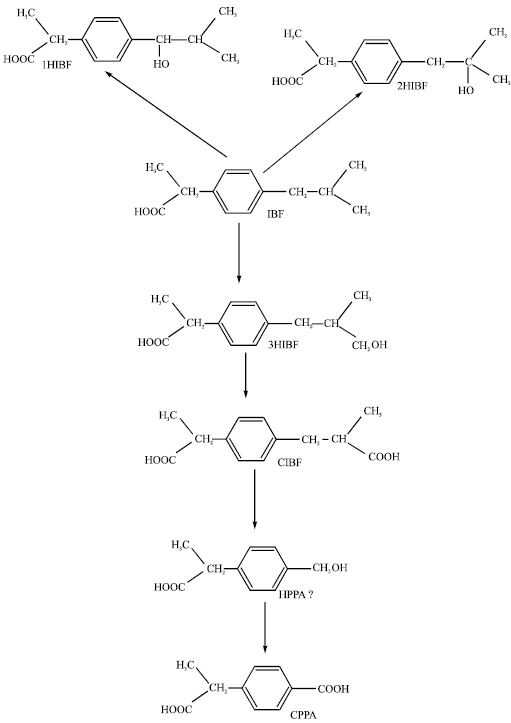 | |
| Fig. 1: | Metabolic pathways for ibuprofen (Tan et al., 2002) - HPPA has not been detected |
MATERIALS AND METHODS
The geometries of ibuprofen and its metabolites have been optimised based on molecular mechanics, semi-empirical and DFT calculations, using the molecular modelling program Spartan ’02. Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimised structures, single point calculations were carried out to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations: molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases. Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
RESULTS AND DISCUSSION
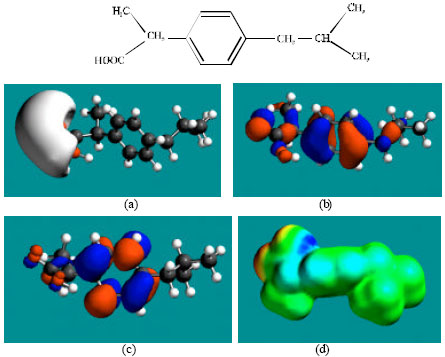
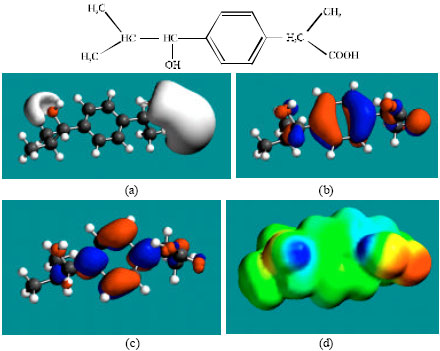
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment, energies of HOMO and LUMO as per both PM3 and DFT calculations for IBF and its metabolites 1HIBF, 2HIBF, 3HIBF, CIBF, HPPA and CPPA. Figure 2-8 give the regions of negative electrostatic potential (greyish-white envelopes) in (a), HOMOs (where red indicates HOMOs with high electron density) in (b), LUMOs in (c), and density of surface charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) as applied to the optimised structures of IBF and its metabolites 1HIBF, 2HIBF, 3HIBF, CIBF, HPPA and CPPA.
The calculated solvation energies of IBF and its metabolites 1HIBF, 2HIBF, 3HIBF, CIBF, HPPA and CPPA from PM3 calculations in kcal mol-1 are, respectively -7.99, -10.55, -10.68, -13.41, -19.18, -14.89 and -19.23 and their dipole moments from DFT calculations are 4.9, 3.5, 6.5, 6.0, 4.2. 4.8 and 2.8, respectively. The values suggest metabolites of IBF would be more soluble in water than the parent drug so that they are more readily excreted via urine. CIBF and CPPA would be most soluble in water and hence most easily excreted.
IBF and its metabolites are found to have large LUMO-HOMO energy differences (that range from 5.50 to 6.61 eV from DFT calculations), indicating that IBF and all its metabolites would be kinetically inert. The relative ease in excretion via urine and the kinetic inertness may explain why the toxic side effects from IBF therapy are found to be low.
In the case of IBF, CIBF and CPPA, the electrostatic potential is found to be more negative around carboxyl oxygen atoms, indicating that the positions may be subject to electrophilic attacks. In the case of 1HIBF, 2HIBF, 3HIBF and HPPA, the electrostatic potential is found to be more negative around carboxyl and hydroxyl oxygen atoms, once again indicating that the positions may be subject to electrophilic attacks.
| Table 1: | Calculated thermodynamic and other parameters of ibuprofen and its metabolites |
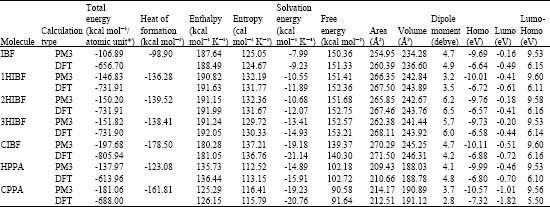 | |
| * in atomic units from DFT calculations | |
 | |
| Fig. 2: | Structure of INH giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
 | |
| Fig. 3: | Structure of NAINH giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
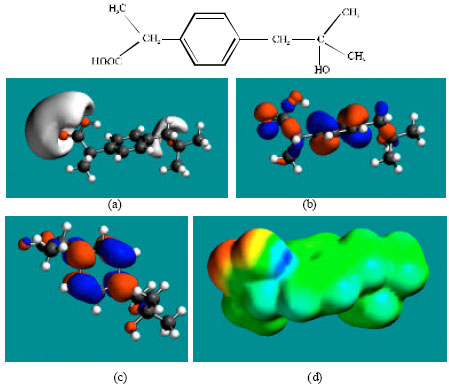 | |
| Fig. 4: | Structure of AcHD giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
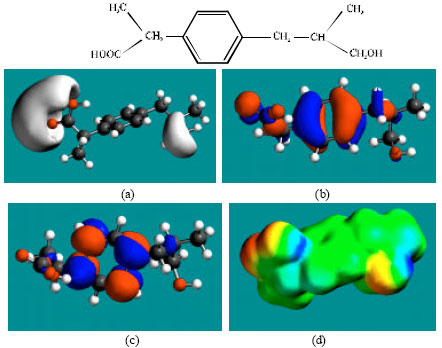 | |
| Fig. 5: | Structure of HD giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
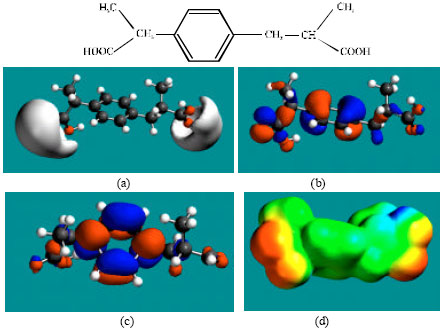 | |
| Fig. 6: | Structure of DAHD giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
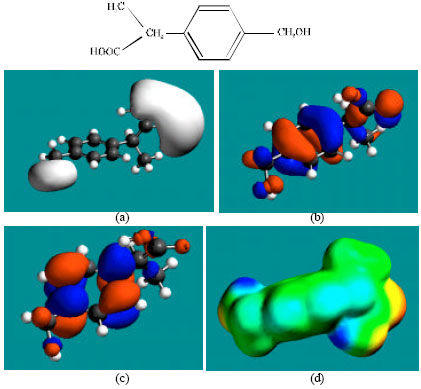 | |
| Fig. 7: | Structure of PH giving the electrostatic potential in (a), HOMOs in (b), LUMOs in (c) and surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) |
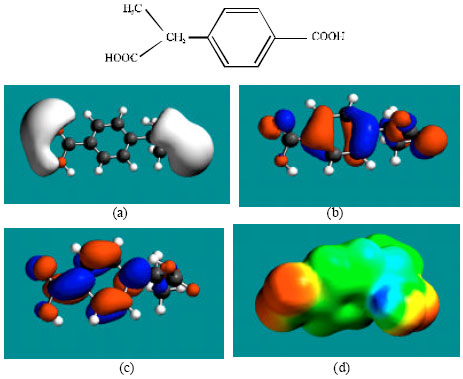 | |
| Fig. 8: | Structure of CPPA giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) |
In the case of IBF, 1HIBF, 2HIBF and 3HIBF, HOMOs with high electron density are found close to most of the non-hydrogen atoms except the two methyl carbon atoms of the isobutyl moiety whereas the LUMOs are found to concentrate on the non-hydrogen atoms of propanoic acid moiety and phenyl ring. In the case of CIBF, HPPA and CPPA both HOMOs with high electron density and LUMOs are found close to most of the non-hydrogen atoms.
The overlap or close proximity of positions of HOMOs with high electron density and those of negative electrostatic potential give further support to the idea that the positions may be subject to electrophilic attacks. When the electron densities on molecular surfaces (Fig. 2d to 8d) are considered, it can be seen that the molecular surfaces of IBF and all its metabolites, have both electron-rich and electron-deficient regions so that they may be subject to both electrophilic and nucleophilic attacks. The latter attack means that IBF and its metabolites can react with cellular glutathione and can oxidize nucleobases in DNA. Reaction with glutathione introduces oxidative stress by compromising the anti-oxidant status of the cell whereas oxidation of nucleobses in DNA causes DNA damage. However, as stated earlier IBF and its metabolites are expected to be kinetically inert so that the rates of such adverse reactions would be low.
CONCLUSIONS
Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that there are some electron-deficient regions on the molecular surfaces of IBF and its metabolites so that they can react with cellular glutathione, thus causing glutathione depletion and hence oxidative stress, and can also cause oxidation of nucleobases in DNA and hence DNA damage. However, the large LUMO-HOMO energy differences observed for IBF and all its metabolites may mean that the rates of such adverse reactions may be low. Thus the mild toxicity associated with the drug IBF may be explained in terms of kinetic inertness and ease in excretion.
ACKNOWLEDGMENTS
The author is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Hao, H., G. Wang and J. Sun, 2005. Enantioselective pharmacokinetics of ibuprofen and involved mechanisms. Drug Metabol. Rev., 1: 215-234.
PubMed - Rorarius, M.G., P. Suominen, G.A. Baer, O. Romppanen and R. Tuimala, 1993. Diclofenac and ketoprofen for pain treatment after elective caesarean section. Br. J. Anaesth., 70: 293-297.
PubMed - Tan, S.C., B.K. Patel, S.H.D. Jackson, C.G. Swift and A.J. Hutt, 2002. Stereoselectivity of ibuprofen metabolism and pharmacokinetics following the administration of the racemate to healthy volunteers. Br. J. Anaesth., 32: 683-697.
CrossRef