
S.N. Depamede
Microbiology and Biotechnology Laboratory, Faculty of Animal Sciences, Mataram University, Jl. Majapahit No. 62, Mataram, NTB, 83125, Indonesia
I. Menz
School of Biological Sciences, Flinders University of South Australia, GPO Box 2100, Adelaide 5001, South Australia
Research Journal of Microbiology
Year: 2011 | Volume: 6 | Issue: 7 | Page No.: 599-608







ABSTRACT
Unlike most mammalian cells, Plasmodium sp., are unable to utilize preformed pyrimidine bases and nucleosides hence they are reliant solely on de novo pathway. Aspartate transcarbamoylase (ATCase, EC 2.1.3.2) catalyzes the first committed step in de novo pyrimidine biosynthesis pathway, is a potential target for novel anti-parasitic including antimalarial drugs. P. falciparum ATCase has not been studied extensively. To reveal whether it has a regulatory subunit or no and how its evolution, phylogenetic analysis and protein modeling of ATCase P. falciparum were studied. The structural model can be used to identify the possible differences between active sites of mammalian and Plasmodium enzyme. This is important in a relation with antimalarial drug development. Analogous sequences from P. falciparum were searched by ‘tBLASTn search’ carried out using the web based tools provided by National Center for Biotechnology Information (NCBI). After alignment of ATCase residues sequences, phylogram was constructed by means of MEGA 2.1 software. The results show that the residues sequences of P. falciparum ATCase in the phylogenetic tree constructed clearly positioned P. falciparum as Class C or “A” consistent with Wild and Wales structural organization. In addition the models for the three dimensional protein structures of the catalytic domain of human and P. falciparum ATCase were also generated. As far as our concerned, our study was the first to reveal of P. falciparum ATCase classification based on its ATCase amino acid sequences. However, the structure of P. falciparum ATCase needs to be determined experimentally to confirm this and to assist the rational design of antimalarial drugs.
PDF Abstract XML References Citation
Received: April 14, 2011;
Accepted: June 03, 2011;
Published: July 29, 2011
How to cite this article
S.N. Depamede and I. Menz, 2011. Phylogenetic Analysis and Protein Modeling of Plasmodium falciparum Aspartate Transcarbamoylase (ATCase). Research Journal of Microbiology, 6: 599-608.
URL: https://scialert.net/abstract/?doi=jm.2011.599.608
URL: https://scialert.net/abstract/?doi=jm.2011.599.608
INTRODUCTION
The synthesis of pyrimidine nucleotides in pathogenic as well as cancer cells is one of the essential targets for novel anti-viral or anti-parasitic (Ridley, 2002), as well as anti-cell proliferative drugs (Galmarini et al., 2003; Striepen et al., 2004). Aspartate transcarbamylase (ATCase) is one of the targets, since ATCase is an enzyme that plays an important role in the first committed step in series of reactions that leads to synthesis of pyrimidine nucleotides (Purcarea et al., 2003; Labedan et al., 2004).
The functional organisation of the enzyme activities is significantly different in prokaryotes, protists, fungi, plants and animals (Nara et al., 2000; Roos, 2005; Ginger, 2006). Prokaryotic ATCases have been classified into three classes (A, B and C) based on their molecular weights, subunit architectures and regulatory responses while the eukaryotes ATCases studied fall into either class “A” or the multifunctional polypeptides such as CA and CAD (Wild and Wales, 1990). Santiago and West (2003, 2008) showed that the ATCase can be used for the analysis of taxonomic diversity within the genus Pseudomonas. E. coli ATCase has both catalytic and regulatory chains which are encoded by pyrB and pyrl genes, respectively reviewed by Helmstaedt et al. (2001). ATCase classification in some parasitic protozoa, such as plasmodium is still unclear (Nara et al., 2000).
In the present study, Plasmodium falciparum ATCase was chosen as a model since Plasmodium have been the serious parasitic protozoa that causes of malaria diseases and P. falciparum is the species that causes most human deaths especially in Asia and Africa (Ursos and Roepe, 2002; Osamor, 2010). As mentioned above, ATCase classification in Plasmodium is still unclear whether the ATCase is composed of catalytic and regulatory polypeptides or if these functions are found on the same polypeptide or if the catalytic chain is unregulated. However, the sequence of a polypeptide with homology to the catalytic subunit has been reported in Plasmodium falciparum and submitted to the EMBL/GenBank/DDBJ databases (Hillier, 2001) with Primary Accession Number O15804. For these reasons, study on P. falciparum ATCase needs to be carried out.
In this study, it was hypothesised that ATCase genes with similar sequences will display similar types of regulation. In order to assign the Plasmodium enzyme to a particular class and thereby predict if the Plasmodium enzyme may have a regulatory subunit or not, phylogenetic analysis of the ATCase amino acid sequence from a variety of species was conducted.
Furthermore, it is becoming apparent that Plasmodium falciparum increasingly resistant to antimalarial drugs, particularly chloroquine which then in turn result in increased morbidity and mortality especially among children (Umar et al., 2007, 2008). It is necessary to develop alternative antimalarial drugs pursued with a clear mechanism of Plasmodium inhibition without disrupting the host’s essential metabolic pathway (Ridley, 2002).
In order to elucidate whether the protein structure of human ATCase has similarity to that of Plasmodium falciparum, a model for the three dimensional protein structures of the catalytic domain of human and P. falciparum ATCase were generated. It was anticipated that these structural models may be able to explain differences observed in the kinetic analysis of the mammalian and Plasmodium enzymes in the future. This should be important in a relation with antimalarial drug development.
MATERIALS AND METHODS
The bioinformatics operations: The bioinformatics operations were carried out mainly using the “BioManager” interface provided by ANGGIS (http://www.angis.org.au), (ANGIS, 2002). Searching for analogous sequences from P. falciparum by ‘tBLASTn search’ was carried out using the web based tools provided by NCBI (http://www.ncbi.nlm.nih.gov/projects/Malaria).
Data bank retrieval and multiple sequence alignments: Genes of E. coli which encoded the catalytic (pyrB) and regulatory (pyrl) subunits of ATCase were used as queries to retrieve all ATCase sequence present in the SwissProt and SpTrEMBL data bases (as of November 2010). Multiple protein sequence alignments were generated using Clustasl W (accurate) program (Thompson et al., 1994).
Phylogenetic tree construction: Phylogenetic tree constructed by MEGA 2.1 software (GENETYX-WIN 5.1) after alignment of ATCase sequences (Tamura et al., 2007).
Protein modeling: Models for three dimensional structures of catalytic domain of human and P. falciparum ATCase were generated either using the automated protein modeling package Swiss-Model (Peitsch, 1996) directly or obtaining the model from the Swiss-Model repository (http://swissmodel.expasy.org/repository/). In all cases the models were based on the E. coli structure (PDB id ld09).
Model structures and the E. coli structure complexed with the bisubstrate analog N-phosphonacetyl aspartate, PALA (Krause et al. 1987), were aligned using the ‘lsqkab’ program which is part of CCP4 suite of programs, Collaborative Computational Project (Winn et al., 2002; Krissinel et al., 2004).
Residues, identified as crucial for the E.coli ATCase catalysis according to Helmstaedt et al. (2001) and Macol et al. (2001) i.e., Ser-52, Thr-53, Arg-54, Thr-55, Arg-105, His-134, Gln-137 and Arg-167 were used for alignment. The active sites of the aligned structures were analysed and diagrams were constructed using Phyton-enhanced molecular graphics program, PyMOL software, DeLano Scientific, California, USA (DeLano, 2002).
RESULTS
The catalytic domain: Text searches were used to find the gene sequences from a variety of organisms, for the ATCase catalytic subunit or domain (pyrB) as presented in material and methods. The results show that 124 mono-functional ATCase genes were retrieved. In addition the sequence for the trifunctional CAD protein complex was identified for a further 93 eukaryotic organisms. The sequence corresponding to the pyrB gene from Plasmodium falciparum was located by text searching with the ATCase EC number (2.1.3.2), this resulted in a single hit: SwissProt+SpTrEMBLO15804.
The regulatory domain: A similar strategy to that used to find the catalytic gene sequences was used to find genes encoding the regulatory subunit (pyrl). The regulatory gene sequences for 39 organisms were found. Text searching failed to identify any P. falciparum genes that are annotated as ATCase regulatory subunits (pyrl).
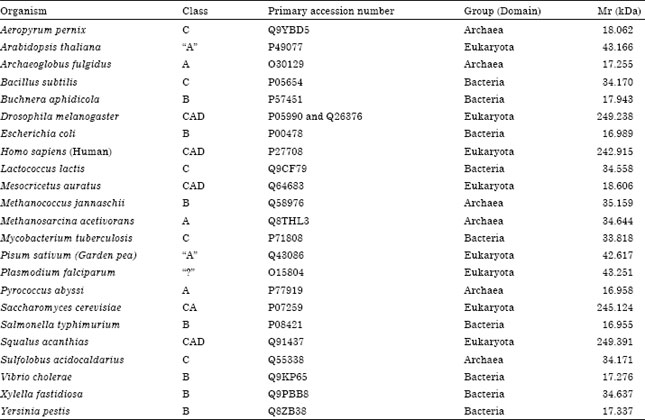
Classification of ATCase: Selected ATCase sequences which were representative of a diverse array of organisms were manually classified based on their size and structural organization according to the classification system of Wild and Wales (1990). The results of this classification are presented in Table 1. It appears here that the ATCase of P. falciparum seems rather difficult to put into which classes among the Wild and Wales’s system. For these reasons it was performed alignment of amino acids and then proceeded with the constructing of a dendogram (Fig. 1).
Alignment and phylogenetic tree: Figure 1 shows the alignment of the amino acid sequences of the ATCase catalytic genes (pyrB) for all the representative organisms used in the manual classification presented in Table 1. A phylogenetic tree from the aligned sequences was derived and the result is presented in Fig. 2. With this construction the position of P. falciparum ATCase became more clear that it was among eukaryote class "A" and archea class C.
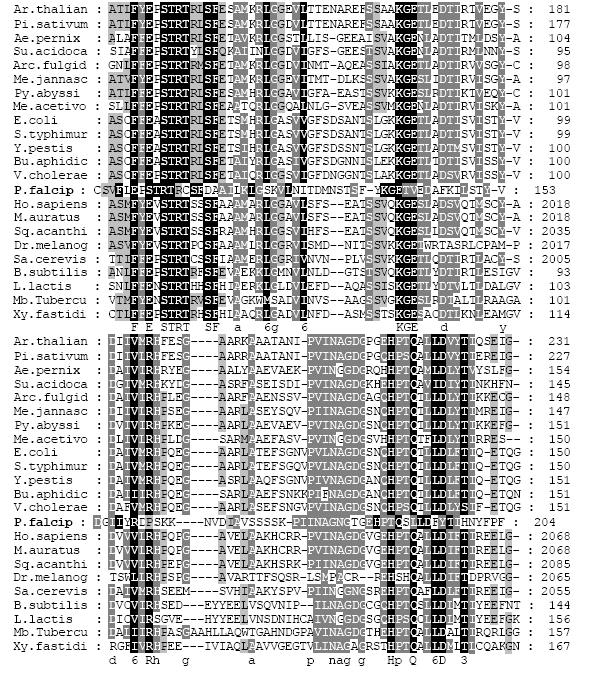 | |
| Fig. 1: | Representative of multiple alignment of aspartate transcarbamoylase catalytic domains of organisms presented in Table 1. Completely conserved resuidues and patially conserved residues are shaded black and grey, respectively |
Protein modeling: The models for the three dimensional protein structures of the catalytic domain of human and P. falciparum ATCase were generated based on the sequence alignment of P. falciparum, the Human and E. coli (Fig. 3). Figure 3 shows the alignment revealed only 20% identity of P. falciparum ATCase to E. coli and human ATCase. Four amino acids, serine, threonine, arginine and threonine (S-107, T-108, R-109 and T-110) were used in PALA binding as presented in three dimensional structures (Fig. 4).
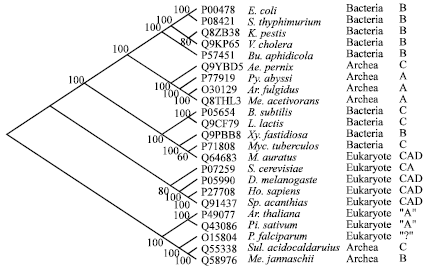 | |
| Fig. 2: | The phylogenetic tree of asparatate transcarbamoylase sequences. The dendogram was constructed using the program Protpars as described in text |
 | |
| Fig. 3: | Alignment of P. Falciparum, E. Coli and Human ATCase catalytic residues. Homologous residues are represented by does. Residues proposed to be involved in PALA binding are shaded grey (STRT, Ser-107, Thr-108, Arg-108 and Thr-110). The positions of the resuidues are numbered after P. Falciparum ATCase sequence |
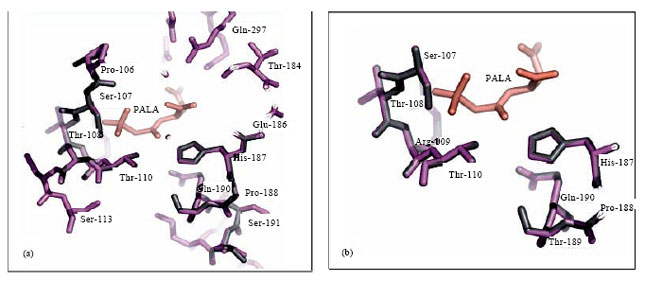 | |
| Fig. 4 (a-b): | Comparison of P. Flaciparum and the Human ATCase active sites. Panel (a) pf-ATCase (purple) and the Human CAD-ATCase (grey) were superimposed as described in text. Panel (b)as in (a) but focusing on the catalytic residues by removing the non catalytic residues. The bisubstrates analogue PALA is shown (salmon) |
| Table 1: | Grouping of representation retrieved ATCase sequences based on ATCase structural organisation modified from Wild and Wales (1990) |
 | |
DISCUSSION
The sequence corresponding to the catalytic domain of the pyrB gene from P. falciparum was located by text searching with the ATCase EC number 2.1.3.2, a single hit of SwissProt+SpTrEMBLO15804 was found. However when a similar strategy was used to find genes encoding the regulatory subunit (pyrl), text searching failed to identify any P. falciparum genes that are annotated as ATCase regulatory sub units (pyrl). To convince the results, BLAST searches of the P. falciparum genome using the E. coli pyrl gene was conducted. Again this approach was also failed to identify any possible pyrl genes for P. falciparum. Hence, it can be assumed that either P. falciparum does not contain a pryl gene homolog or the homology between P. falciparum and E. coli are very low. Therefore, it is likely that the P. falciparum ATCase does not contain a regulatory subunit but this cannot be conclusively ruled out.
The constructed phylogenetic tree (Fig. 2) in general groups the organisms in a similar manner as the classification systems proposed by Bethell and Jones (1969) and Wild and Wales (1990), who grouped the bacteria into three classes A, B and C. This is especially true for the general grouping of prokaryotes and eukaryotes.
Figure 2 shows that the phylogenetic approach also tends to group the prokaryotes into three categories which correspond to those of as determined by enzyme morphology. However, the phylogenetic approach does not completely resolve the morphologically derived ATCase subclasses for the prokaryotic organisms (Labedan et al., 2004).
A similar result is also seen for the eukaryotic groups. The phylogenetic tree shows a clear eukaryotic grouping within ATCase classes (Fig. 2).
In animals, the ATCase exists as a domain of multifunctional enzyme recognized as CAD (Hemmens and Carrey, 1995). In the present study the CAD groups were found together with the CA class which is the ATCase class for yeast. This is in agreement with the classification of Wild and Wales (1990) who grouped yeast and hamster ATCase into classes CA and “A”, respectively. Based on the molecular weight, class A ATCases are the biggest but these are the least comprehensively studied. The bifunctional enzyme complexes (CA) containing both ATCase and Carbamoyl Phosphatase (CPSase). Class C ATCase are much smaller and lack a regulatory domain or subunit (Vickrey et al., 2002; Labedan et al., 2004).
Interestingly P. falciparum and the plants Pisum sativum and Arabidopsis thaliana group distinctly from the other eukaryotic sequences (Fig. 2). The P. falciparum sequence sits between the eukaryotic and prokaryotic (class B) groupings making it difficult to determine which group it is more similar to. A similar phylogenetic study by Nara et al. (2000) also had difficulties assigning the P. falciparum ATCase sequence to the prokaryote or eukaryote groupings.
The reason why P. falciparum does not group with the eukaryotes such as D. melanogaster, is likely due to the fact that P. falciparum is the only eukaryote reported with a monofunctional ATCase (Hillier, 2001). This is in line with strong evidence that the three gene (pyr1, pyr2, pyr3) organization in the P. falciparum genome are not clustered (Krungkrai et al., 2003).
The present study suggests that P. falciparum ATCase belongs to a subclass of its own which falls between the B and CAD of the classification system of Wild and Wales (1990). It was anticipated that these structural models may be able to explain differences observed in the kinetic analysis of the mammalian and Plasmodium enzymes reported elsewhere in this study. Models of the three dimensional protein structures of the catalytic domain of human and P. falciparum ATCase were generated as described in methods based on the sequence alignment of P. falciparum, the human and E. coli.
As shown in the multiple alignments presented in Fig. 1 and 3 shows 20% highly conserved residues among the three species. This is especially true of the residues have been identified as crucial for catalysis such as those equivalent to Ser-52, Thr-53, Arg-54, His-134 and Gln-137 in E. coli (as reviewed by Helmstaedt et al. (2001), Macol et al. (1999) and Macol et al. (2001).
The α-carbon position of the residues which define the catalytic site were used to superimpose the two modelled structures with the E. coli structure complexed with the bisubstrate analogue N-(phosphonacetyl) aspartate, PALA (Krause et al. 1987). The superposition of the active site residues (Pro-106, Arg-107, Thr-108, Arg-109, Thr-110, His-187, Pro-188, Thr-189 and Gln-190) are shown in Fig. 4. It is evident, in these models that there is very little difference in the position of residues that are capable of interacting with PALA (<3Å) suggesting that the architecture of the active site is extremely conserved. However, one must be cautious when making this conclusion as both models have been constructed using the same template which may bias their similarity to the E. coli PALA binding site (<3Å). PALA has been well known as a potent ATCase inhibitor (Boxstael et al., 2003) however, Purcarea et al. (2003) revealed that PALA was a relatively ineffective inhibitor of CPSase-ATCase complex. Purcarea et al. (2003) carried out the study on Aquifex aeolicus ATCase. Whether similar characteristics are also found in P. falciparum ATCase, need to be proven further. Hence, further studies investigating the structural biology of P. falciparum ATCase are still needed.
CONCLUSION
The residues sequences of P. falciparum ATCase in the phylogenetic tree constructed clearly positioned P. falciparum as Class C or “A” based on structural organization of Wild and Wales (1990). Structural models constructed shown a strong similarity between P. falciparum ATCase and the human ATCase. However, the structure of P. falciparum ATCase needs to be determined experimentally to confirm this and to assist the rational design of antimalarial drugs.
ACKNOWLEDGMENT
This research work (SND) was funded in part by Department of Higher Education, Ministry of National Education Republic of Indonesia and Asian Development Bank (ADB)-TPSDP No. 1792-INO.
REFERENCES
- Boxstael, S.V., R. Cunin, S. Khan and D. Maes, 2003. Aspartate transcarbamylase from the hyperthermophilic archaeon Pyrococcus abyssi: Thermostability and 1.8 Å resolution crystal structure of the catalytic subunit complexed with the bisubstrate analogue N-phosphonacetyl-L-aspartate. J. Mol. Biol., 326: 203-216.
CrossRef - Galmarini, C.M., K. Kamath, A. Vanier-Viornery, V. Hervieu and E. Peiller, et al., 2003. Drug resistance associated with loss of p53 involves extensive alterations in microtubule composition and dynamics. Br. J. Cancer, 88: 1793-1799.
CrossRef - Ginger, M.L., 2006. Niche metabolism in parasitic protozoa. Phil. Trans. R. Soc. B., 361: 101-118.
CrossRef - Helmstaedt, K., S. Krappmann, and G.H. Braus, 2001. Allosteric regulation of catalytic activity: escerichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiol. Mol. Biol. Rev., 65: 404-421.
CrossRef - Hemmens, B., and E.A. Carrey, 1995. Mammalian dihydroorotase secondary structure and interactions with other proteolytic fragments from the multienzyme polypeptide CAD. Eur. J. Biochem., 231: 220-225.
CrossRef - Krause, K.L., K.W. Volz and W.N. Lipscomb, 1987. 2.5 A structure of aspartate carbamoyltransferase complexed with the bisubstrate analog N-(phosphonacetyl)-L-aspartate. J. Mol. Biol., 193: 527-553.
Direct Link - Krissinel, E.B., M.D. Winn, C.C. Ballard, A.W. Ashton and P. Patel, et al., 2004. The new CCP4 Coordinate Library as a toolkit for the design of coordinate-related applications in protein crystallography. Acta Cryst., 60: 2250-2255.
CrossRef - Krungkrai, J., P. Prapunwatana, C. Wichitkul, S. Reungprapavut, S.R. Krungkrai and T. Horii, 2003. Molecular biology and biochemistry of malarial parasite pyrimidine biosynthetic pathway. South East Asian J. Trop. Med. Public Health, 34: 32-43.
PubMedDirect Link - Macol, C., M. Dutta, B. Stec, H. Tsuruta and E.R. Kantrowitz, 1999. The 80s loop of the catalytic chain of Escherichia coli aspartate transcarbamoylase is critical for catalysis and homotropic cooperativity. Protein Sci., 8: 1305-1313.
CrossRef - Macol, C.P., H. Tsuruta, B. Stec and E.R. Kantrowitz, 2001. Direct structural evidence for a concerted allosteric transition in Escherichia coli aspartate transcarbamoylase. Nature Struct. Biol., 8: 423-426.
CrossRef - Nara, T., T. Hshimoto and T. Aoki, 2000. Evolutionary implications of the mosaic pyrimidine-biosynthetic pathway in eukaryotes. Gene, 257: 209-222.
PubMedDirect Link - Osamor, V.C., 2010. The etiology of malaria scourge: A comparative study of endemic nations of Africa and Asia. J. Biol. Sci., 10: 440-447.
CrossRefDirect Link - Peitsch, M.C., 1996. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans., 24: 274-279.
PubMed - Ridley, R.G., 2002. Medical need, scientific opportunity and the drive for antimalarial drug. Nature, 415: 686-693.
CrossRef - Santiago, M.F. and T.P. West, 2003. Comparison of aspartate transcarbamoylase regulation in Pseudomonas alcaligenes and Pseudomonas mendocina. J. Basic Microbiol., 43: 75-79.
Direct Link - Santiago, M.F. and T.P. West, 2008. Regulation of aspartate transcarbamoylase activity in Pseudomonas putida biovar B. Res. J. Microbiol., 3: 331-335.
CrossRefDirect Link - Striepen, B., A.J.P. Pruijssers, J. Huang, C. Li and M.J. Gubbels et al., 2004. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci., 101: 3154-3159.
CrossRef - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Thompson, J.D., D.G. Higgins and T.J. Gibson, 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22: 4673-4680.
CrossRefPubMedDirect Link - Umar, R.A., N.M. Jiya, S.W. Hassan, K. Abdullahi, J.M. Ahmed and U. Nataala, 2007. Apparent drug failure following artesunate treatment of Plasmodium falciparum malaria in sokoto, Nigeria: Three case reports. Trends Med. Res., 2: 113-116.
CrossRefDirect Link - Umar, R.A., S.W. Hassan, M.J. Ladan and M.N. Jiya et al., 2008. Therapeutic efficacy of chloroquine for uncomplicated Plasmodium falciparum malaria in nigeria children at the time of transition to artemisinin based combination therapy. Res. J. Parasitol., 3: 32-39.
CrossRefDirect Link - Vickrey, J.F., G. Herve and D.R. Evans, 2002. Pseudomonas aeruginosa aspartate transcarbamoylase: Characterization of its catalytic and regulatory properties. J. Biol. Chem., 277: 24490-24498.
Direct Link - Wild, J.R. and M.E. Wales, 1990. Molecular evolution and genetic engineering of protein domains involving aspartate transcarbamoylase. Annu. Rev. Microbiol., 44: 193-218.
CrossRef - Winn, M.D., A.W. Ashton, P.J. Briggs, C.C. Ballard, and P. Patel, 2002. Ongoing developments in CCP4 for high-throughput structure determination. Acta Cryst., 58: 1929-1936.
PubMed
florence mactouff Reply
What are the major structural differences between Plasmodium falciparum ATCASE and human host enzyme.