
Abdulazeez T. Lawal
Nano Science and Sensor Technology Research Group, School of Applied Sciences and Engineering, Monash University, Churchill, Vic-3842, Australia
Samuel B. Adeloju
Nano Science and Sensor Technology Research Group, School of Applied Sciences and Engineering, Monash University, Churchill, Vic-3842, Australia
Journal of Applied Sciences
Year: 2012 | Volume: 12 | Issue: 4 | Page No.: 315-325







ABSTRACT
The preparation of two electrochemical (potentiometric and amperometric) phosphate biosensors is described and compared. Purine Nucleoside Phosphorylase (PNP) and xanthine oxidase (XOD) were co-immobilised via entrapment into polypyrrole (PPy) films by galvanostatic polymerization. Polypyrrole entrapment was achieved with 0.5 M pyrrole by using a polymerisation time of 200 sec and a mole ratio of 1:8 (6.2 U mL-1 XOD: 49.6 U mL-1 PNP) in both biosensors. Sensitive amperometric measurements were compared with those of potentiometric measurements obtained for PPy-PNP-XOD-Fe(CN)64¯ biosensors. A minimum detectable concentration of 1.0 μM phosphate and a linear concentration range of 5-20 μM were achieved with potentiometric PPy-PNP-XOD-Fe(CN)64¯ biosensor. In comparison, a minimum detectable concentration of 10 μM and a linear concentration range of 0.1-1 mM were achieved with amperometric biosensor. The presence of uric and ascorbic acids had the least effect on the performance of the amperometric and potentiometric PPy-PNP-XOD-Fe(CN)64¯ biosensors, therefore will not have any effect on phosphate measurement in both biosensors at levels normally present in water.
PDF Abstract XML References Citation
Received: December 01, 2011;
Accepted: January 24, 2012;
Published: March 21, 2012
How to cite this article
Abdulazeez T. Lawal and Samuel B. Adeloju, 2012. Polypyrrole Based Amperometric and Potentiometric Phosphate Biosensors: A Comparative Study. Journal of Applied Sciences, 12: 315-325.
DOI: 10.3923/jas.2012.315.325
URL: https://scialert.net/abstract/?doi=jas.2012.315.325
DOI: 10.3923/jas.2012.315.325
URL: https://scialert.net/abstract/?doi=jas.2012.315.325
INTRODUCTION
Phosphate’s widespread presence in detergent and fertiliser has made its determination of great importance to environmental pollution control. High phosphate concentration can pollute water resources and causes eutrophication of lakes and rivers (Nakamura, 2010). The eutrophication of water by phosphate can lead to over-growth of plant and toxic algae, thereby making it unsuitable for drinking or industrial use. In food analysis, the adverse effect of excess intake of phosphate in food additives upon human health is now a major consideration and is of vital interest to researchers (Nordin, 1997). Phosphate determination is also important in clinical diagnosis; the determination of phosphate in body fluid provides useful information about certain diseases and about the energetic state of cells and bone function (Kivlehan et al., 2009; Shervedani and Pourbeyram, 2009).
Phosphate determination based on spectroscopy (Galhardo and Masini, 2000; Mckelvie, 2000; Li et al., 2002; Nakamura, 2004; Yaqoob et al., 2004; Lin et al., 2006; Okoh et al., 2006; Gimbert et al., 2007; Nevesa et al., 2008) and chromatographic techniques (Galceran et al., 1993; Bello and Gonzalez, 1996; Colina et al., 1996; Zhu et al., 2008) were the analytical techniques commonly used for phosphate site monitoring. However, these required the use of hazardous chemicals for sample pre-treatment which are time consuming. They also produced toxic waste, such as tartaric acid and heavy metal such as antimony. Ion selective electrodes were also used for phosphate determination, based on various metals and associated complexes such as Sn complexes (Chaniotakis et al., 1993; Sasaki et al., 2004), hydroxyapatite (Petrucelli et al., 1996) cobalt metallic wires (Chen et al., 1997, 1998; De Marco and Phan, 2003; Gimbert et al., 2007; Bai et al., 2011). Comparatively these electrodes suffered from low selectivity and poor stability of the ion selective membranes. Other methods used for determination of phosphate included flow injection systems based on luminescence (Kyung and Hoon, 2009; Andolina and Morrow, 2011; Cardemil et al., 2010), chemiluminescence (Kawasaki et al., 1989; Nakamura et al., 1999; Nakamuraac et al., 2003; Yaqoob et al., 2004; Motomisu and Li, 2005) and florescence reactions (Motomisu and Li, 2005; Wang et al., 2010), conductometry (Zhang et al., 2008) and screen printed electrodes (Kwan et al., 2005; De Albuquerque and Ferreira, 2007; Zou et al., 2007; Khaled et al., 2008).
A simple alternative is the use of an enzyme sensor that is able to measure phosphate directly in the sample. Enzyme sensors have been developed based on enzymatic sequences in which a first enzyme (usually a phosphorylase) uses phosphate as a co-substrate giving a product that is the substrate for a second enzyme, usually an oxidase. Among these enzyme sensors are phosphate biosensors that use, as biorecognition elements, substances such as nucleoside phosphorylase and xanthine oxidase (D’Urso and Coulet, 1990, 1993; Male and Luong, 1991; Wollenberger et al., 1992; Su and Mascini, 1995; Chen et al., 1997; Vazquez et al., 2003) and most commonly used alkaline phosphatase (Adeloju and Lawal, 2011). Others are acid phosphatase (Guilbault and Nanjo, 1975; Guilbault, 1984), glucose oxidase (Su and Mascini, 1995; Zhang et al., 2008), pyruvate oxidase (Mori and Kogure, 1994; Ikebukuroa et al., 1996; Mak et al., 2003; Roger et al., 2005; Rahman et al., 2006; Akylmaz and Yorgancia, 2007), sucrose phosphorylase (Adeloju and Lawal, 2011), phosphoglucomutase and glucose 6-phosphate dehydrogenase (Adeloju and Lawal, 2011). Combination of Maltose Phosphorylase (MP), mutarotase (MR) and glucose oxidase (GOX) have recently been used for fabrication of phosphate biosensors (Mousty et al., 2001; Zhang et al., 2008).
Electrochemical biosensor has made the production of screen printed electrode easier and amenable to miniaturisation of biosensors (Roger et al., 2005; De Albuquerque and Ferreira, 2007; Zou et al., 2007; Khaled et al., 2008). This has facilitated its mass production for quick and quantitative phosphate site monitoring or evaluation.
Amperometric biosensor requires three electrodes and application of potential before measurable current can be obtained. The use of enzyme-based amperometric biosensor has increased considerably in the past ten years as a result of its high selectivity and the sensitivity of amperometric signal (Mak et al., 2003; De Albuquerque and Ferreira, 2007; Lawal and Adeloju, 2009a). There are few potentiometric biosensors that require simple construction of two electrodes and without the application of potentials for measurable potential signal to be generated (Adeloju and Lawal, 2005; Lawal and Adeloju, 2009a; Adeloju and Lawal, 2011). Amperometric sensing can introduce interference as a result of oxidation of other matrix components which can lead to erroneous and enhanced current signal. However, in potentiometric sensing, oxidation of other matrix components are avoided). Villalba et al. (2009) recently reviewed the advantages and disadvantages of the electrochemical biosensors for the determination of phosphate.
In the PNP-XOD bienzyme system employed recently in various studies (Cosnier et al., 1998; Adeloju and Lawal, 2005; Lawal and Adeloju, 2009b; Lawal and Adeloju, 2009a; Adeloju and Lawal, 2011) showed a higher amount of hypoxanthine was produced during enzymatic phosphate recycling. Enzymatic phosphate recycling also took place using MP/GOX/Ap trienzyme for low level phosphate detection (Conrath et al., 1995; Huwel et al., 1997; Mousty et al., 2001). Wollenberger (Wollenberger et al., 1992) employed amplification by enzymatic substrate recycling in order to lower the detection limit, involving co-immobilisation of alkaline phosphatase (aP) and glucose oxidase. In the presence of phosphate ion inosine was phosphorylated by PNP to ribose-1-phosphate. Phosphate was then liberated by aP catalysis and became available again for inosine phosphorylation. Phosphate was thus recycled between aP and PNP while a higher amount of hypoxanthine was produced and recognised by sequential oxidation by XOD Hypoxanthine was subsequently oxidised to H2O2 catalysed by XOD (Watanabe et al., 1987; D’Urso and Coulet, 1990, 1993; Wollenberger et al., 1992), as given in Eq. 1 and 2.
| (1) |
and:
| (2) |
In this study, biosensors were developed based on the enzymatic reaction shown in Eq. 1 and 2.
The successful immobilisation of XOD into polypyrrole film by Lawal and Adeloju (2009a, b, 2010) demonstrates that co-immobilisation with PNP is feasible. To this end, the comparison of electrochemical phosphate PPy-PNP-XOD biosensors in current study involved investigations of the effect of enzyme (PNP:XOD) mole ratio, pH, inosine and buffer concentrations, analytical characteristic, stability and common interferences affecting the two electrochemical biosensors.
MATERIALS AND METHODS
Reagents and standard solutions: Xanthine oxidase (XOD) (EC.1.2.3.2.2 Grade1 (2.0 U mg-1) from buttermilk, purine nucleoside phosphorylase (PNP) (EC.2.4.2.1.2 (15 U mg-1), potassium ferrocyanide and pyrrole were obtained from Sigma. Pyrrole was distilled under vacuum at 130°C prior to use and this was stored in an aluminium foil-covered sample bottle in the freezer to prevent UV degradation until required for use. K4Fe (CN)6 also undergoes UV degradation and so the solution was stored until required. XOD stock was stored in the refrigerator and PNP was stored in the freezer until required. All chemicals used were reagent grade and all compounds used in this work were prepared without further purification.
Instrumentation: A potentiostat/galvanostat designed and constructed in our laboratories was used for electrochemical measurements the potentiostat/ galvanostat was used in galvanostatic mode for the electropolymerisation. A three-electrode cell, which contained platinum working electrode, a platinum wire counter electrode and a Ag/AgCl (3 M KCl), was employed for amperometric detection of phosphate while potentiometric measurements were performed in a two electrode cell. The potentiostat was connected to a computer controller system. Solution was stirred when necessary with a Sybron Thermolyne (model S-17410) stirrer.
Enzyme immobilisation
Electrode preparation: A 320 μm aluminium oxide powder was used to polish the platinum working electrode with a soft polishing pad, to remove any previous film and then finally polished with 5 μm aluminium oxide. The electrode surface was washed thoroughly with Milli-Q water, rinsed under a stream of acetone and finally rinsed thoroughly with Milli-Q to remove any of the remaining aluminium oxide. The electrode was dried with fibre-free tissue paper and fixed onto a retort stand for the next step.
Electropolymerisation of PPy-PNP-XOD film: A three-electrode cell, which contained platinum working electrode, a platinum wire counter electrode and an Ag/AgCl (3 M KCl), was employed for electropolymerisation of PPy film. PPy-PNP-XOD-Fe(CN)64- biosensor, was made by immobilisation of xanthine oxidase (6.2 U mL-1), purine nucleoside phosphorylase (48 unit mL-1) and 50 mM potassium ferrocyanide (K4Fe (CN)6) into a polypyrrole film. The electropolymerisation was accomplished in the presence of pyrrole monomer (0.1-0.5 M) at a chosen current density (0.125-0.5 mA cm-2) and polymerisation time of 200 sec. The polymer electrode formed after galvanostatic polymerisation, was washed several times under a stream of Milli-Q water to remove any weakly bound XOD or PNP or K4Fe(CN)6 molecules prior to electrochemical measurements.
Amperometric measurements: An Ag/AgCl (3 M KCl) reference electrode, a platinum wire auxiliary electrode and PPy-PNP-XOD-Fe (CN)64- working electrode were assembled into a 20 mL voltametric cell. The supporting electrolyte used for the amperometric measurements was a 0.05 M barbitone buffer solution (pH 7.8) which contained 0.1 M sodium chloride and 10 mM inosine. The solution in the electrochemical cell was stirred with a magnetic stirrer. A potential of 0.2 V was then applied to the electrode and the current response to the addition of a phosphate standard solution was recorded.
Potentiometric measurements: The electrode was rinsed thoroughly with distilled water to remove any loosely bound enzyme after electropolymerisation. Phosphate measurement was performed by placing the electrode into a magnetically stirred 20 mL (0.05 M) barbitone buffer solution, which contained 0.1 M NaCl and 5 mM inosine. The resulting equilibrium potential versus Ag/AgCl electrode was then measured after each addition of standard phosphate solution in a two-electrode cell. The interference of uric acid, ascorbic acid and glycine on the potentiometric responses were tested by their addition into the cell prior to the potentiometric measurements.
RESULTS AND DISCUSSION
Response to phosphate: In amperometric biosensor, XOD in the presence of molecular oxygen oxidised Hx and produced hydrogen peroxide which was detected by PPy-PNP-XOD-Fe(CN)64- biosensor. The reduced mediator was simultaneously regenerated on the electrode surface giving an amperometric signal directly proportional to phosphate concentration. But with potentiometric biosensor, a potential sensing signal was developed and the potential difference developed may have originated from the redox couple of hydrogen peroxide produced. Figure 1 shows a typical amperometric response of the amperometric biosensor upon the successive addition of phosphate into electrochemical cell with stirring.
Effect of PNP and XOD ratio: The performance of biosensors for environmental monitoring in terms of detection limit, stability and calibration range were strictly dependent on the enzyme loading. Figure 2 shows the results obtained for varying the XOD:PNP ratio in the outer layer. Both biosensors show that the optimum response was obtained when a mole ratio between 1:8 and 1:10 of XOD:PNP was incorporated into the outer layer. This is very similar to the mole ratio reported by D’Urso and Coulet (1990, 1993) for a phosphate biosensor.
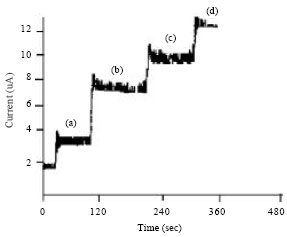 | |
| Fig. 1: | Typical amperometric response of amperometric PPy-PNP-XOD-Fe(CN)64- biosensor to phosphate. (a) 10, (b) 20, (c) 30 and (d) 40 mM phosphate. The monomer solution contained 0.4 M pyrrole, current density: 0.5 mA cm-2 polymerisation period: 200 sec |
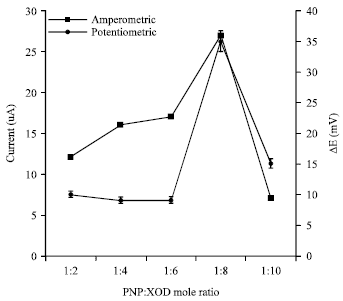 | |
| Fig. 2: | Effect of immobilised (PNP) and (XOD) on phosphate response with both amperometric and potentiometric PPy-PNP-XOD-Fe(CN)64- biosensors. (XOD) was 6.2 U mL-1, Fe(CN)64- was 50 mM and (Phosphate) was 10 mM. The monomer solution contained 0.4 M pyrrole, current density: 0.5 mA cm-2 polymerisation period: 200 sec |
Guilbault and Cserfalvi (1976) reported a ratio of 1:10, while Kulys et al. (1992) and Wollenberger et al. (1992) reported a ratio of 1:5 for their construction of an amperometric phosphate biosensor.
 | |
| Fig. 3: | Effect of pH on phosphate response obtained with both amperometric and potentiometric PPy-PNP-XOD- Fe(CN)64- biosensors. [Phosphate] was 10 mM. The monomer solution contained 0.4 M pyrrole, 6.2 U mL-1 of XOD, current density: 0.5 mA cm-2 polymerisation period: 200 sec and 50 mM Fe(CN)64- |
On the other hand (Yao et al., 2003) reported an optimum ratio of 1:21, while (Konisita et al., 1995) found a ratio of 1:3 to be the optimum ratio for their amperometric phosphate biosensor.
Effects of pH and inosine concentration: Figure 3 shows the effect of pH on the response of phosphate on both amperometric and potentiometric PPy-PNP-XOD biosensors. The optimum pH of the sensors was found to be between 7 and 8 close to the optima of the two enzymes in solution (Berger and Grass, 1989; Guilbault and Nanjo, 1975; Kulys et al., 1992) found that pH 7 produced the optimum sensitivity for an amperometric phosphate biosensor. While D’Urso and Coutlet (1990) and Yao et al. (2003) found pH 7.5 to be ideal for their amperometric phosphate biosensor. No response to phosphate was obtained when either one or both PNP and XOD were not included in the immobilisation or if the sample did not contain inosine, which had to be supplied in excess in a phosphate-free buffer. Wollenberger used excess inosine to ensure a co-reactant independent of phosphate response was obtained. Figure 4 illustrates the influence of inosine concentration on the phosphate response in both amperometric and potentiometric biosensors.
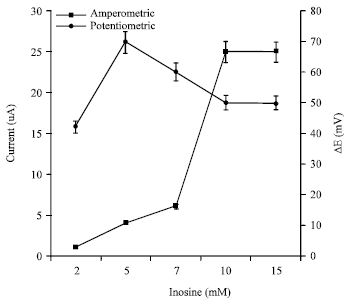 | |
| Fig. 4: | Effect of inosine concentration on phosphate response obtained with both amperometric and potentiometric PPy-PNP-XOD- Fe(CN)64- biosensors. (Phosphate) was 10 mM. The monomer solution contained 0.4 M pyrrole, 6.2 U mL-1 of XOD, current density: 0.5 mA cm-2 polymerisation period: 200 sec and 50 mM Fe(CN)64- |
The chosen inosine concentration of 10 mM was optimum for obtaining the most sensitive current response for phosphate in amperometric biosensor. Also further increase in inosine concentration did not raise the sensitivity of the phosphate response. For potentiometric biosensor, on the other hand 5 mM inosine was found to be sufficient. D’Urso and Coulet used 0.1 mM of inosine, while Used 5 mM of inosine in 0.05 M borate buffer. Wollenberger used 0.09 mom of inosine. Although, the amount of inosine used by Watanabe was not specified, it was in excess in 0.05 M Tris-HCl buffer solution. Guilbault used inosine concentration of 5 mM in 0.05 M tris-HCl buffer at pH 7.0. The optimum inosine concentration of 10 mM established in amperometric biosensor was higher, possibly due to the application of high voltage that could degrade inosine that lowers the electrochemical response. The amount used here was still close to the levels used in some studies.
Effect of buffer concentration: H2O2 and uric acid were produced when PNP catalysed the phosphorylysis of inosine in presence of phosphate to ribose-1-phosphate and hypoxanthine. The H2O2 and uric acid were electrochemically active and therefore could be easily detected. A signal could be obtained due to change in pH caused by the production of uric acid and optimisation of the buffer concentration was therefore essential. The influence of the concentration of buffer solution on the biosensors response is shown in Fig. 5.
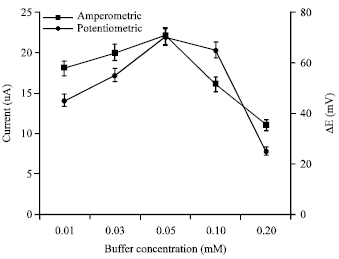 | |
| Fig. 5: | Effect of buffer concentration on phosphate response obtained with both amperometric and potentiometric _PPy-PNP-XOD-Fe (CN)64- biosensors. (Phosphate) was 10 mM. The monomer solution contained 0.4 M pyrrole, 6.2 U mL-1 of XOD, current density: 0.5 mA cm-2 polymerisation period: 200 sec and 50 mM Fe(CN)64- |
In amperometric biosensor a buffer concentration of 0.05 M produced optimum phosphate response. At lower concentrations the current continued to increase, whereas at higher concentrations the current gradually decreased. Higher buffering capacity at higher concentrations of buffer due to its high ionic strength affected phosphate current response; this in turn hindered the movement of H2O2 to the electrode. While in potentiometric biosensor the increasing buffer concentration initially reduced the potentiometric response and then increased with decreasing buffer concentration up to 0.05 M as in amperometric biosensor. A weak buffer was therefore necessary to enable adequate measurement of the potentiometric response, because the higher buffering capacity of the more concentrated buffer solution affected the magnitude of the response. The optimum buffer concentration of 0.05 M was used for both amperometric and potentiometric biosensors.
Interference characteristics: Ascorbic Acid (AA) is considered to be a major interferant to most biosensors because of its relatively high concentrations in biological samples and low oxidation potential (Vidal et al., 2000). Figure 5 shows that the presence of ascorbic acid of up to 1 mM did not interfere with the phosphate response. However, the data shows that the addition of 5 mM ascorbic acid resulted in the enhancement of the phosphate response by 28%. The effect of AA at higher concentration on the response is even more dramatic.
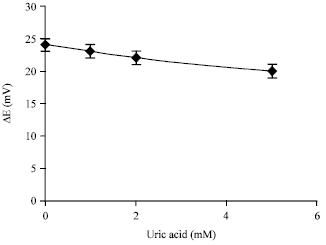 | |
| Fig. 6: | Effect of uric acid on the potentiometric response of phosphate obtained with BSA-GLA-XOD-PNP biosensor. Potentiometric measurement was made in 0.05 M tris -HCl buffer. (Phosphate) was 10 mM. XOD:PNP ratio was 1:8 |
This enhancement effect of AA is very similar to those reported for other phosphate and glucose biosensors that employ the same enzyme immobilisation method (Su and Mascini, 1995; Adeloju and Moline, 2001) However, it is unlikely that the presence of low concentrations of ascorbic acid in water will affect the performance of the phosphate biosensor when used for analysis of water samples. AA concentration greater than 2.5 mM enhanced the response by up to 42%.
Another common interferant is uric acid but it is not as limiting as ascorbic acid (Su and Mascini, 1995; Adeloju and Moline, 2001). The presence of 1 mM uric acid suppressed the potentiometric response of the biosensor by 4%. It has been found in previous study (Kulys et al., 1992) that the sensitivity of the enzyme-based biosensors is degraded by the presence of uric acid. Smooth baselines and well defined responses were obtained for concentrations of uric acid less than 1 mM. When PPy-PNP-XOD-Fe(CN)64- biosensor fabricated by enzyme immobilisation in polypyrrole film was used, the presence of 2.5 mM uric acid the change in potential was suppressed by 17% as shown in Fig. 6. The presence of a high concentration of uric acid in water or other aqueous solutions favours the oxidation of uric acid with hydrogen peroxide, thus suppressing the electrochemical response of the phosphate. Interference of AA with phosphate sensor may be due to oxidation of ascorbic acid by the H2O2 produced from the enzymatic reaction. Adeloju and Moline (Adeloju and Moline, 2001) also found that ascorbic acid and uric acid affected their potentiometric PPy film sensor. The presence of glycine in water or other aqueous solutions did not interfere with both biosensors’ response to phosphate and it is probable that other proteins would not interfere either.
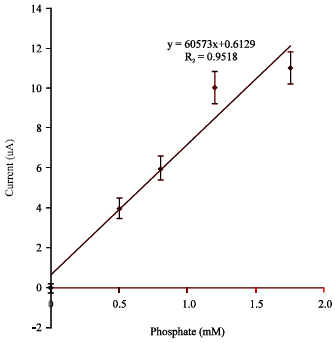 | |
| Fig. 7: | Typical calibration graph obtained for phosphate with amperometric PPy-PNP-XOD-Fe(CN)64- biosensor. Phosphate was 10 mM. The monomer solution contained 0.4 M pyrrole, 6.2 U mL-1 of XOD, current density: 0.5 mA cm-2 polymerisation period: 200 sec, 200 mV and 50 mM Fe(CN)64-Eapp.200 mV |
However, the performance of both PPy-PNP-XOD-Fe(CN)64- biosensors fabricated by enzyme immobilisation in polypyrrole film for the determination of phosphate in water samples will not be affected by the presence of low concentrations of uric acid.
Analytical characteristics: Figure 7 shows a typical calibration curve obtained for amperometric phosphate biosensor (PNP-XOD-Fe(CN)64-. The amperometric response was linear for phosphate concentrations between 0.1 and 1 mM. The minimum detectable phosphate concentration with the biosensor was 10 μM. (1.0 mg) On the other hand, the calibration curve obtained for potentiometric biosensor is as shown in Fig. 8. The potentiometric response was linear between 5 and 25 μM (0.5-2.5 mg L-1) and the minimum detectable phosphate concentration was 1 μM (0.1 mg L-1) which is much lower than the minimum obtained in amperometric biosensor. It is also lower than the limit of detection for phosphate using AP/GOX combination (4 μM) (Villalba et al., 2009), pyruvate biosensor of Kwan-Roger (3.6 μM) (Roger et al., 2005) and is also comparable to those obtained with photometric molybdite complex (0.3 μM) (Villalba et al., 2009) or sensitive fluorescence methods (0.8 μM) (Villamil-Ramos and Yatsimirsky, 2011).
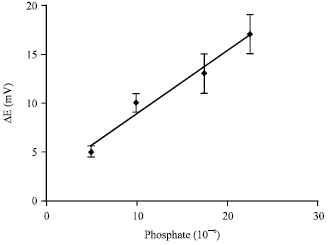 | |
| Fig. 8: | Typical calibration graph obtained for phosphate with potentiometric PPy-PNP-XOD-Fe(CN)64- biosensor. (Phosphate) was 10 mM. The monomer solution contained 0.4 M pyrrole, 6.2 U mL-1 of XOD, current density: 0.5 mA cm-2 polymerisation period: 200 sec and 50 mM Fe(CN)64- |
Potentiometric biosensor has the same minimum detection limit (0.1 μM) as that of conductometry biosensor produced by Zhang et al. (2008). This is enough to probe phosphate concentration within inland water ways. A plot of the potential versus logarithm of phosphate concentration produced a slope of 46.5±1.0 mV/decade which was higher than the expected 29.5 mV/decade for a two-electron process. However, this value was similar to a slope of 45.5 mV/decade reported previously by Menzel for a phosphate biosensor (Menzel and Lerch, 1995). The observed linear relationship between changes in potential and phosphate concentration, instead of log concentration, may be due to the complex nature of the composite electrode and the extent to which the Nernstian behaviour is maintained, which is also dependent on various parameters, such as enzyme loading, film thickness and substrate (phosphate ion). Further work is necessary to improve the sensitivity of these biosensors to permit direct determination of phosphate concentrations in water, as these are still not sensitive enough for phosphate analysis in potable water.
Stability of biosensor response: Figure 9 shows that the phosphate response obtained with the amperometric PPy-PNP-XOD-Fe(CN)64- decreased slowly by 20% of its initial value after 24 h. In general, the PPy-PNP-XOD-Fe(CN)64- electrode lost more than 20% of its sensitivity per day if it was not stored in barbitone buffer at pH between 7 and 7.8.
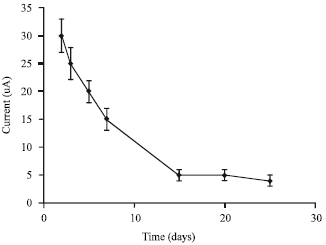 | |
| Fig. 9: | Influence of storage time on the sensitivity of phosphate response obtained with amperometric PPy-PNP-XOD-Fe(CN)64- biosensor. Amperometric measurement was performed under the following conditions: 0.05 M barbitone buffer, applied potential of 200 mV and 10 mM inosine. Phosphate was 10 mM |
The amperometric response of the biosensor decreased rapidly for the first two days and at eighth day it had reached 50% of its initial value. Beyond 12 days it continued to decrease and eventually stabilised at about 20% of its initial value after two weeks. In potentiometric biosensor, the response started to decrease after two weeks of continuous use. It maintained 80% of its initial potentiometric response in the first three weeks but after four weeks there was a decrease of 60% of it initial potentiometric response. The observed decrease in electrochemical responses of the biosensors may be attributed to the loss of enzyme and ferrocyanide into the bulk solution and also possibly be to lifetime of the enzymes.
In conductometry phosphate biosensor produced by Zhang et al. (2008), 70% of the initial response was lost after two weeks and 50% after three weeks. Male and Luong have reported that the use of the same bienzyme system on reactivated nylon membrane resulted in a loss of 30% of its response in three weeks (Male and Luong, 1991). The lower loss observed in Male and Louong (1991) study may be attributed to its use in the FIA mode, where the electrode was in direct contact with the analyte for an only relatively short period. Despite the 20% decrease in sensitivity observed in amperometric biosensor, the resulting response is still useful for quantification of phosphate by standard addition methods. However, when the PPy-PNP-XOD-Fe(CN)64- electrode is stored in a barbitone buffer solution the stability of enzymes is improved, optimum sensitivity is attained and analysis of phosphate ion is improved.
CONCLUSIONS
Amperometric and potentiometric PPy-PNP-XOD-Fe(CN)64- biosensors have been fabricated for accurate potentiometric and amperometric measurements of phosphate at concentrations that are suitable for environmental monitoring. Potentiometric biosensor can detect a minimum of 1 μM phosphate ion and has a linear concentration range of 5-20 μM, where as amperometric biosensor can detect a minimum of 10 μM and has a linear range of 0.1-1 mM. Interferences from uric and ascorbic acids, at levels normally present, were not considered problematic for detection of phosphate with these biosensors. The biosensors appear promising for analysing phosphate in polluted water. However, further improvement in sensitivity was necessary before these biosensors are reliably used in unpolluted and portable waters.
REFERENCES
- Adeloju, S.B. and A. Lawal, 2005. Polypyrrole-based bilayer biosensor for potentiometric determination of phosphate in natural waters. Int. J. Environ. Chem., 85: 771-780.
CrossRef - Adeloju, S.B.O. and A.T. Lawal, 2011. Fabrication of a bilayer potentiometric phosphate biosensor by cross-link immobilization with bovine serum albumin and glutaraldehyde. Anal. Chimi. Acta, 691: 89-94.
PubMed - Adeloju, S.B. and A.N. Moline, 2001. Fabrication of ultra-thin polypyrrole-glucose oxidase film from supporting electrolyte-free monomer solution for potentiometric biosensing of glucose. Biosens. Bioelect., 16: 133-139.
Direct Link - Akyilmaz, E. and E. Yorganci, 2007. Construction of an amperometric pyruvate oxidase enzyme electrode for determination of pyruvate and phosphate. Electrochim. Acta, 52: 7972-7977.
CrossRefDirect Link - Andolina, C.M. and J.R. Morrow, 2011. Luminescence resonance energy transfer in heterodinuclear lnIII complexes for sensing biologically relevant anions. Eur. J. Inorgan. Chem., 2011: 154-164.
CrossRef - Bai, Y., J.H. Tong, C. Bian and S.H. Xia, 2011. Fabrication and characterization of cobalt nanostructure-based microelectrodes for phosphate detection. Key Eng. Mater., 483: 559-564.
CrossRef - Bello, M.A. and A.G. Gonzalez, 1996. Determination of phosphate in cola beverages using nonsuppressed ion chromatography: an experiment introducing ion chromatography for quantitative analysis. J. Chem. Educ., 73: 1174-1176.
CrossRefDirect Link - Cardemil, C.V., D.R. Smulski, R.A. Larossa and A.C. Vollmer, 2010. Bioluminescent Escherichia coli strains for the quantitative detection of phosphate and ammonia in coastal and suburban watersheds. DNA Cell Biol., 29: 519-531.
PubMed - Chaniotakis, N.A., K. Jurkschat and A. Ruhlemann, 1993. Potentiometric phosphate selective electrode based on a multidendate-tin(IV) carrier. Anal. Chim. Acta, 282: 345-352.
Direct Link - Chen, Z., R. de Marco and P.W. Alexander, 1997. Flow-injection potentiometric detection of phosphates using ametallic cobalt wire ion-selective electrode. Anal. Commun., 34: 93-95.
CrossRefDirect Link - Chen, Z., P. Grierson and M.A. Adams, 1998. Direct determination of phosphate in soil extracts by potentiometric flow injection using a cobalt wire electrode. Anal. Chim. Acta, 363: 191-197.
CrossRef - Colina, M., H. Ledo, E. Gutierrez, E. Villalobos and J. Marin, 1996. Determination of total phosphorus in sediments by means of high pressure bombs and ion chromatography. J. Chromatogr. A, 739: 223-227.
CrossRefDirect Link - Conrath, N., B. Grundig, S. Huwel and K. Cammann, 1995. A novel enzyme sensor for the determination of inorganic phosphate. Anal. Chim. Acta, 309: 47-52.
Direct Link - Cosnier, S., C. Gondran, J.C. Watelet, W. De Giovani, R.P.M. Furriel and F.A. Leone, 1998. A bienzyme electrode (Alkaline Phosphatase-Polyphenol Oxidase) for the amperometric determination of phosphate. Anal. Chem., 70: 3952-3956.
CrossRef - D'Urso, E.M. and P. Coulet, 1990. Phosphate-sensitive enzyme electrode: A potential sensor for environmental control. Anal. Chim. Acta, 239: 1-5.
Direct Link - De Marco, R. and C. Phan, 2003. Determination of phosphate in hydroponic nutrient solutions using flow injectio potentiometry and a cobalt-wire phophate ion selective electrode. Talanta, 60: 1215-1221.
Direct Link - D'Urso, E.M. and P.R. Coulet, 1993. Effect of enzyme ratio and enzyme loading on the performance of a bienzymatic electrochemical phosphate biosensor. Anal. Chim. Acta, 281: 535-542.
Direct Link - Galceran, M.T., M. Diez and L. Paniagua, 1993. Determination of phosphate in samples with high levels of sulphate by ion chromatography. J. Chromatogr. A, 657: 77-85.
CrossRefDirect Link - Galhardo, C.X. and J.C. Masini, 2000. Spectrophotometric determination of phosphate and silicate by sequential injection using molybdenum blue chemistry. Anal. Chim. Acta, 417: 191-200.
CrossRefDirect Link - Guilbault, G.G. and T. Cserfalvi, 1976. Ion selective electrodes for phosphate using enzyme systems. Anal. Lett., 9: 277-289.
CrossRef - Guilbault, G.G. and M. Nanjo, 1975. A phosphate-selective electrode based on immobilized alkaline phosphatase and glucose oxidase. Anal. Chim. Acta, 78: 69-74.
CrossRefDirect Link - Huwel, S., L. Haalck, N. Conrath and F. Spener, 1997. Maltose phosphorylase from Lactobacillus brevis: Purification, characterization and application in a biosensor for ortho-phosphate. Enzyme Microb. Technol., 21: 413-420.
PubMed - Ikebukuroa, K., R. Nishida, H. Yamamoto, Y. Arikawa and H. Nakamura et al., 1996. A novel biosensor system for the determination of phosphate. J. Biotechnol., 48: 67-72.
Direct Link - Kawasaki, H., K. Sato, J. Ogawa, Y. Hasegawa and H. Yuki, 1989. Determination of inorganic phosphate by flow injection method with immobilized enzymes and chemiluminescence detection. Anal. Biochem., 182: 366-370.
PubMed - Khaled, E., H. Hassan, A. Girgis and R. Metelka, 2008. Construction of novel simple phosphate screen-printed and carbon paste ion-selective electrodes. Talanta, 77: 737-743.
CrossRef - Kivlehan, F., W.J. Mace, H.A. Moynihanb and D.W.M. Arrigana, 2009. Study of electrochemical phosphate sensing systems: Spectrometric, potentiometric and voltammetric evaluation. Electrochimica Acta, 54: 1919-1924.
CrossRef - Konisita, H., D. Yoshidaa, K. Miki, T. Usui and T. Ikeda, 1995. An amperometric-enzymatic method for assays of inorganic phosphate and adenosine deaminase in serum based on the measurement of uric acid with a dialysis membrane-covered carbon electrode. Anal. Chim. Acta, 303: 301-307.
Direct Link - Kulys, J., I.J. Higgins and J.V. Bannister, 1992. Amperometric dertermination of phosphate ions by biosensor. Biosens. Bioelect., 7: 187-191.
CrossRefDirect Link - Kwan, R.C.H., H.F. Leung, P.Y. Hon, H.C. Cheung, K. Hirota and R. Renneberg, 2005. Amperometric biosensor for determining human salivary phosphate. Analytical Biochem., 343: 263-267.
PubMed - Lawal, A.T. and S.B. Adeloju, 2009. Development of a polypyrrole-based amperometric phosphate biosensor. J. Applied Sci., 10: 1907-1914.
CrossRef - Lawal, A.T. and S.B. Adeloju, 2010. Polypyrrole-based potentiometric phosphate biosensor. J. Mol. Catalysis, 63: 45-49.
CrossRef - Li, Y., Y. Muo and H.M. Xie, 2002. Simultaneous determination of silicate and phosphate in boiler water at power plants based on series flow cells by using flow injection spectrophotometry. Anal. Chim. Acta, 455: 315-325.
Direct Link - Lin, X., X. Wu, Z. Xie and K.Y. Wong, 2006. PVC matrix membrane sensor for fluorescent determination of phosphate. Talanta, 70: 32-36.
Direct Link - Mak, W.C., C. Chan, J. Barford and R. Renneberg, 2003. Biosensor for rapid phosphate monitoring in a sequencing batch reactor (SBR) system. Biosensors Bioelect., 19: 233-237.
Direct Link - Male, K.B. and J.H.T. Luong, 1991. An FIA biosensor system for the determination of phosphate. Biosens. Bioelect., 6: 581-587.
CrossRefDirect Link - Motomisu, S. and Z.H. Li, 2005. Trace and ultratrace analysis methods for the determination of phosphorus by flow-injection techniques. Talanta, 66: 332-340.
Direct Link - Mousty, C., S. Cosnier, D. Shan and S. Mu, 2001. Trienzymatic biosensor for the determination of inorganic phosphate. Anal. Chim. Acta, 443: 1-8.
Direct Link - Nakamuraac, H., M. Hasegawab, Y. Nomuraac, K. Ikebukuroad, Y. Arikawab and I. Karubea, 2003. Improvement of a CL-FIA system using maltose phosphorylase for the determination of phosphate-ion in freshwater 1. Analytical Lett., 36: 1805-1817.
CrossRef - Nakamura, H., H. Tanaka, M. Hasegawa, Y. Masuda and Y Arikawa et al., 1999. An automatic flow-injection analysis system for determining phosphate ion in river water using pyruvate oxidase G (from Aerococcus viridans). Talanta, 50: 799-807.
PubMedDirect Link - Nevesa, M.S.A.C., M.R.S. Soutob, I.V. Toth, S.M.A. Victal, M.C. Drumond and AOSS Rangel, 2008. Spectrophotometric flow system using vanadomolybdophosphate detection chemistry and a liquid waveguide capillary cell for the determination of phosphate with improved sensitivity in surface and ground water samples. Talanta, 77: 527-532.
Direct Link - Okoh, M.P., J.L. Hunter, J.E.T. Corrie and M.R. Webb, 2006. A biosensor for inorganicphosphate using Rodamine labneled phosphate bidng protein. Biochemistry, 45: 14764-14771.
Direct Link - Petrucelli, G.C., E.Y. Kawachi, L.T. Kubota and C.A. Bertran, 1996. Hydroxyapatite-based electrode: A new sensor for phosphate. Anal. Commun., 33: 227-229.
Direct Link - Rahman, M.A., D.S. Park, S.C. Chang, C.J. Mcneil and Y.B. Shim, 2006. The biosensor based on the pyruvate oxidase modified conducting polymer for phosphate ions determinations. Biosens. Bioelect., 21: 1116-1124.
Direct Link - Roger, C.H.K., H.F. Leung, P.Y.T. Hon, J.P. Barford and R. Renneberg, 2005. A screen-printed biosensor using pyruvate oxidase for rapid determination of phosphate in synthetic wastewater. Applied Microbiol. Biotechnol., 66: 377-383.
CrossRefDirect Link - Sasaki, S., S. Ozawa, D. Citterio, K. Yamada and K. Suzuki, 2004. Biosensor for rapid phosphate monitoring in a Sequencing Batch Reactor (SBR) system. Talanta, 63: 131-134.
CrossRefDirect Link - Shervedani, R.K. and S. Pourbeyram, 2009. Zirconium immobilized on gold-mercaptopropionic acid self-assembled monolayer for trace determination of phosphate in blood serum by using CV, EIS and OSWV. Biosensors Bioelectron., 24: 2199-2204.
CrossRefDirect Link - Su, Y. and M. Mascini, 1995. AP-GOD biosensor based on a modified poly(phenol) film electrode and its application in the determination of low levels of phosphate. Anal. Lett., 28: 1359-1378.
CrossRefDirect Link - De Albuquerque, Y.D. and L.F. Ferreira, 2007. Amperometric biosensing of carbamate and organophosphate pesticides utilizing screen-printed tyrosinase-modified electrodes. Anal. Chem. Acta, 596: 210-221.
PubMed - Vazquez, M.J., B. Rodriguez, C. Zapatero and D.G. Tew, 2003. Determination of phosphate in nanomolar range by an enzyme-coupling fluorescent method. Anal. Biochem., 320: 292-298.
PubMedDirect Link - Vidal, J.C., E. Garcia-Ruiz and J.R. Castillo, 2000. Strategies for the improvement of an amperometric cholesteror biosensor based on electropolymerisation in flow system: Use of charge transfer mediator and platinisation of electrode. J. Pharm. Biomed. Anal., 24: 51-62.
CrossRefPubMedDirect Link - Villalba, M.M., K.J. McKeegan, D.H. Vaughan, M.F. Cardosi and J. Davis, 2009. Bioelectroanalytical determination of phosphate: A review. J. Mol. Catal. B: Enzym., 59: 1-8.
Direct Link - Villamil-Ramos, R. and A.K. Yatsimirsky, 2011. Selective fluorometric detection of pyrophosphate by interaction with alizarin red S-dimethyltin(iv) complex. Chem. Commun., 47: 2694-2696.
CrossRefDirect Link - Wang, D., X. Zhang, C. He and C. Duan, 2010. Aminonaphthalimide-based imidazolium podands for turn-on fluorescence sensing of nucleoside polyphosphates. Org. Biomol. Chem., 8: 2923-2925.
CrossRefDirect Link - Watanabe, E., H. Endo and K. Toyama, 1987. Determination of phosphate Ions with an enzyme sensor system. Biosensors, 3: 297-306.
Direct Link - Wollenberger, U., F. Schubert and F.W. Scheller, 1992. Biosensor for sensitive phosphate detection. Sensors Actuators B: Chem., 7: 412-415.
CrossRef - Yao, T., K. Takashima and Y. Nanjyo, 2003. Simultaneous determination of orthophosphate and total phosphates (inorganic phosphates plus purine nucleotides) using a bioamperometric flow-injection system made up by a 16-way switching valve. Talanta, 60: 845-851.
Direct Link - Yaqoob, M., A. Nabi and P.J. Worsold, 2004. Determination of nanomolar concentrations of phosphate in freshwaters using flow injection with luminol chemiluminescence detection. Anal. Chim. Acta, 510: 213-218.
Direct Link - Zhang, Z., N. Jaffrezic-Renault, F. Bessueille, D. Leonarda and S. Xia et al., 2008. Development of a conductometric phosphate biosensor based on tri-layer maltose phosphorylase composite films. Anal. Chim. Acta, 615: 73-79.
Direct Link - Zhu, M., B. Xie, G. Tang, A. Hu, M. Fang, Z. Wu and Y. Zhao, 2008. Quantitative determination of zidovudine diaryl phosphate triester pro-drugs in rat plasma by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal., 48: 1417-1424.
CrossRefPubMedDirect Link - Zou, Z., J. Han, A. Jang, P.L. Bishop and C.H. Ahn, 2007. A disposable on-chip phosphate sensor with planar cobalt microelectrodes on polymer substrate. Biosens. Bioelectron., 22: 1902-1907.
CrossRefDirect Link - Lawal, A.T. and S.B. Adeloju, 2009. Comparison of methods of immobilisation in phosphate biosensor. Biosensor. Bioelectron., 25: 406-410.
CrossRef