
M. I.D. Helal
Department of Soil Sciences, Faculty of Agriculture, Cairo University, Giza, Egypt
Journal of Agronomy
Year: 2006 | Volume: 5 | Issue: 3 | Page No.: 406-417







ABSTRACT
Surface properties, point of zero salt effect (PZSE) and the remaining charge (σp) at the same pH, cation exchange capacity (CEC), sorption/desorption of P and oxalate sorption, are studied for amorphous Al and Fe hydroxides formed in absence and presence of low (oxalate and citrate) and high (tannate) molecular weight organic ligands. Co-precipitation of organic ligands with Al and Fe significantly shifted PZSE of the precipitation products to lower or higher pH values. As oxalate/(Al or Fe) molar ratio (MR) increased from 0 to 0.2, PZSE of Al precipitates significantly shifted to higher pH values (from 5.65 to 7.9), whereas, those of Fe ones shifted to lower pH values (from 6.4 to 5.95). In case of tannate ligand, PZSE of Al and Fe precipitates markedly shifted to lower pH values (from 5.65 to 3.6 and 6.4 to 2.96, respectively). Also, in the presence of citrate ligand, PZSE of Al and Fe precipitates shifted to lower pH values. In the presence of oxalate ligand, the remaining positive charge (σp) of Al and Fe precipitates increased, whereas, it reversed to be negative in the presence of citrate or tannate ones. Co-precipitation of organic ligands with Al or Fe significantly enhanced CEC values of the precipitation products from 32.9 to 62.4 and 58.9 to 68.0 Cmolc kg-1, pH 5.5, respectively. CEC values increased, as the molecular weight of co-precipitated ligand increased. All Al and Fe precipitation products had high capacity to sorb P (268 to 1413 mmol kg-1). Co-precipitation of oxalate ligand with Al or Fe considerably increased P sorption capacity of the precipitation products, whereas, co-precipitation of citrate or tannate ligands suppress P sorption capacity. A good harmony was observed between P sorption capacities of Al and Fe precipitates and PZSE values. The quantities of P desorbed from the previously sorbed varied widely between the precipitates and increased with sorption density. Fe and Al precipitates formed in the presence of oxalate ligand (Fe-Oxal, Al-Oxal) as well as those formed in absence of organic ligands (Fe-OH and Al-OH), which had a remaining positive charge at PZSE, had high ability to sorb and retain sorbed P. On the other hand, those formed in the presence of tannate or citrate ligands, which had a remaining negative charge, had limited one in this connection. Oxalate sorption capacity of Al and Fe precipitates are markedly lower than P. Co-precipitation of organic ligands with Al or Fe precipitates significantly reduced their capacity to sorb oxalate ions.
PDF Abstract XML References
How to cite this article
M. I.D. Helal, 2006. Surface Properties of Amorphous Al and Fe Hydroxides Formed in the Presence of Oxalate, Citrate and Tannate Ligands. Journal of Agronomy, 5: 406-417.
DOI: 10.3923/ja.2006.406.417
URL: https://scialert.net/abstract/?doi=ja.2006.406.417
DOI: 10.3923/ja.2006.406.417
URL: https://scialert.net/abstract/?doi=ja.2006.406.417
INTRODUCTION
Organic acids are nearly ubiquitous in forest and agricultural soils. A wide variety of low-molecular-weight aliphatic organic acids have been identified, including oxalic, citric, formic, acetic, malic, succinic, malonic, maleic, lactic, acontic and fumaric acids (Stevenson, 1967; Huang and Schnitzer, 1986). Formation of precipitation products of Al and Fe and their reactivity are influenced by organic ligands common in soils and the associated environments (Violante and Violante, 1980; Liu and Huang, 2001). Co-precipitation of organic ligands with Al and Fe perturbs mineral crystallization, promotes formation of poorly crystalline and/or finely crystalline precipitation products of Al and Fe (Violante and Huang, 1984, 1985) and significantly altered their surface properties, specific surface area, point of zero charge (PZC) and the remaining charge (σp) at the same pH (Violante and Huang, 1984; Liu and Huang, 1999; Goldberg et al., 2001). That is besides, their subsequent effect on the reactivity of the precipitates toward adsorption/ desorption and transformation of nutrients and pollutants. The hydrolytic reactions of Al and the surface properties of the precipitation products, are affected by the nature and amount of perturbing ligands (Violante and Jackson, 1979; Violante and Huang, 1985; Liu and Huang, 1999). The presence of citric acid during the formation of Fe oxides significantly altered the fine-scale morphology, surface geometry and other surface characteristics of the products (e.g., specific surface area and point of zero salt effect, PZSE), Liu and Huang (1999). Also, the nature and amount of organic ligands co-precipitated with Al and Fe played a significant role in their reactivity toward anion sorption capacity. The pseudoboehmite sample formed in the presence of aspartic acid retained more P than those formed in the presence of tartaric, citric or tannic acids (Violante and Huang, 1989; Violante and Gianfreda, 1995). The amount of P adsorbed per unit weight of the Fe oxides initially decreased with the increase of citrate concentration from 0 to 10 μM and then steadily increased with increase of citrate concentration to 100 and 1000 μM.
Point of zero charge (PZC), is defined as the pH at which the absolute value of the surface charge becomes equal to zero. It is one of the most important characteristics of variable charge minerals (Kosmulski, 2004; Jara et al., 2005). The surface charge behavior was determined by point of zero charge (PZC) measured by electrophoretic mobility (isoelectric point, IEP) and determined by potentiometeric titration (point of zero salt effect, PZSE), Jara et al. (2005). The pHPZSE for the lowest possible ionic strengths (0.001, 0.01 M) provides a reasonable estimate of the pHPZC (Sverjensky, 2005). Although, PZC does not provide a complete description of the oxides (Strauss et al., 1997), charge data for oxides are usually summarized using the point of zero charge (PZC). The presence of organic ligands during the formation of Al and Fe (hydr)oxides significantly shifted PZC values of the precipitation products. Thereby, the variation on reactivity of metal oxides/hydroxides toward adsorption of anions and metal cations could be attributed to their PZC. PZC values of the pseudoboehmites formed in the presence of inorganic and organic ligands vary from 8.0 to 9.3, whereas PZSE value of iron oxide obtained at various citrate concentrations vary from 3.9 to 7.0. The remaining charge (σp) at pH equals PZC, as defined by Uehara and Gillman (1980) and expressed as the amount of H+ or OH¯ adsorbed at PZC, varied as kind and amount of perturbing ligand varied. The capability of organic and mineral components to adsorb H+ or OH¯ at PZC, are contribute to the value of σp (Sakurai et al., 1988). σp of metal hydroxides vary as the co-precipitated ligand varied. Liu and Huang (2001) found that, Fe oxide formed in the presence of citrate ligand had lower PZSE and more negative charge at the same pH. Cation exchange capacity (CEC) has a marked influence on partitioning of cations between the soil solid phase and the soil solution. Therefore, it is one of the most important soil properties controlling the solubility and bioavailability of trace metals (Bolan et al., 1999).
The objective of this research was to study the effect of the presence of low (oxalate and citrate) and high (tannate) molecular weight organic ligands, during the formation of amorphous Al and Fe hydroxides, on surface properties of the precipitation products. Surface properties, point of zero salt effect (PZSE) and the remaining charge (σp) at the same pH and cation exchange capacity (CEC), were determine for synthetic amorphous Al and Fe hydroxides formed in absence or presence of organic ligands. Also, the subsequent effect of these properties on the reactivity of the precipitates toward sorption/desorption of P and sorption of oxalate, were studied. Amorphous Al and Fe hydroxides were chosen for this research due to their abundant occurrence in an environment contains organic ligands (Cornell and Schwertmann, 1979. That is beside, these materials generally have larger surface area and greater reactivity toward anion adsorption than their correspondings of crystalline mineral phases (Bohn et al., 1979). Citrate and oxalate ligands were chosen because they are very common in soil rhizosphere, with concentrations ranging from 109 to 1000 μM (Robert and Berthelin, 1986) and 25 to 1000 μM (Fox and Comerford, 1990), respectively, whereas, tannate ligand was chosen for comparison between low and high molecular weight organic ligands.
MATERIALS AND METHODS
Preparation of amorphous Al (III) hydroxides: A solution of 0.01 M AlCl3 was titrated in the absence or presence of oxalate, citrate or tannate ligands, with 0.1 M NaOH to pH 7.0. The ligand/Al molar ratio (MR) was 0.1. Oxalate, citrate or tannate ligands were added in the form of oxalic, citric and tannic acid at concentration of 0.001 M. The final suspensions were aged for 24 h in an air-conditioned-room at 23±0.5°C, centrifuged at 10000 rpm and washed with deionized water until free of Cl¯ and then freeze dried. Al precipitation products; either the non-modified (Al-OH) which formed in absence of organic ligands, or those modified by co-precipitation of citrate, oxalate or tannate ones (Al-Cit, Al-Oxal and Al-Tan, respectively), were soluble in both acid ammonium oxalate (pH = 3) and 1M HCl, indicating that there had been little or no crystallization (Kinniburgh et al., 1975).
Using the abovementioned procedure, another forms of Al precipitates were prepared in the presence of different two concentrations (2x10-4 and 2x10-3 M) of oxalate or tannate ligands, i.e., at two different MR of 0.02 and 0.2. These molar ratios (MR) are higher and lower, respectively, than the previous one (0.1).
Preparation of amorphous Fe (III) hydroxides: A stock suspension of iron gel was prepared by rapid neutralization of reagent grade Fe(NO3)3 in absence or presence of oxalate, citrate or tannate ligands, at ligand/Fe MR of 0.1, with NaOH to pH 7.0 in a polypropylene beaker. Weights of 0.297, 0.693 or 5.614 g of oxalic, citric and tannic acid, respectively, were added to 13.332 g of Fe(NO3)3. 9 H2O crystal, and mixed thoroughly. After the suspension had been maintained at pH 7.0 for 1 h, the suspension was made up to the appropriate volume (100 mL) so that the final concentration was 0.33 and 0.033 M with respect to Fe and ligands, respectively. Aging took 30 min in an air conditioned-room at 23±0.5°C. The procedure above mentioned with Al was adopted.
Also another forms of Fe-precipitates were prepared in the presence of oxalate or tannate ligands at two ligand/Fe MR of 0.02 and 0.2. Ligand concentrations of the new forms were 6.6x10-3 and 6.6x10-2 M.
Point of zero salt effect (PZSE) and the remaining charge (σp): Point of zero salt effect (PZSE) and the remaining charge at the same pH (σp), expressed as sorbed H+/OH¯, of Al and Fe precipitation products, were determined using potentiometeric-salt-titration (STPT) method as described by Sakurai et al. (1988). This method is initially described for the determination of PZC. Strictly speaking, PZC determined using this method (Sakurai et al., 1988) corresponds to the PZSE described by Sposito (1984), Choi et al. (1999). The pH values of suspensions of Al and Fe precipitates containing varied concentrations of acid or base were measured pHwater. pHsalt values were obtained after the addition of the electrolyte. Adsorbed H+ and OH¯ were calculated and plotted against pHwater and pHsalt. The intersection point of pHwater and pHsalt corresponds to STPT-PZSE. The remaining charge at PZSE (σp) was obtained based on the amount of H+ or OH¯ adsorbed at the same pH.
Cation exchange capacity (CEC): Cation exchange capacity of Al and Fe precipitates was determined using the method of Gillman (1979). This method is suitable for materials consist of variable charged matrices. In this method, the sample saturated and subsequently equilibrated with Ba using BaCl2 solution and then reacted with MgSO4 to replace Ba with Mg. The loss of Mg from the reactant solution is equivalent to that adsorbed and hence to CEC. Mg concentration in the equilibrium solutions was determined using an Atomic Absorption Spectrophotometer (AA- 6200, Shimadzu, Japan)
Sorption/Desorption of P: To 100 mg Al or Fe precipitation product, 100 mL of 50 mM NaClO4 solution containing 0.0 to 2.7 mMP L-1 in the form of KH2PO4, was added. Using 0.1 N HNO3 or 0.1 N NaOH, pH of NaClO4 and KH2PO4 solution were adjusted to 7.0 or 5.5 prior addition to adsorbents, then readjusted after addition. The suspensions were shaked gently for 24 h at 23±0.5°C in a thermostatically controlled room. The suspensions adjusted repeatedly to pH 7.0 or 5.5 throughout the reaction period. The final suspensions were centrifuged at 10000 rpm for 30 min. P concentration was determined in the supernatant solution by the phosphomolybdate blue method as described by Murphy and Riley (1962) and modified by Watanabe and Olsen (1965). The quantity of sorbed P was calculated from the difference between the determined initial and final concentration of P in equilibrium solution. To all suspensions, a few drops of toluene were initially added to prevent microbial activity. All experiments were done in two replicates. The total number of protons consumed in periodical adjusting of the reacted solutions was calculated.
The previously sorbed P was desorbed using 0.5 M NaHCO3, pH 8.5 at same adsorbent: solution ratio of 1:1000. The suspension was shaked gently for reaction period of 24 h at 23±0.5°C, then centrifuged. P concentration was determined in the supernatant solution as described before.
Oxalate sorption: To 100 mg Al or Fe precipitation product, 100 mL of 50 mM NaClO4 solution, containing 0 to 1.175 mM L-1 oxalate in the form of H2C2O4, was added. The pH of NaClO4 and H2C2O4 solutions were adjusted to pH 7.0 or 5.5 prior addition to adsorbents, then readjusted after addition. The procedure above mentioned with P was adopted. Final oxalate concentration was measured using Ion Chromatography (761 Compact IC, Metrohm).
Statistical analysis: Data of point of zero salt effect (PZSE), cation exchange capacity (CEC), P sorption/desorption and oxalate sorption, were analyzed using linear model procedure of SAS program (SAS, 1996). Except for data of PZSE, the models consists of three factors including; 1) two types of metal hydroxides (Al and Fe), 2) four forms of Al precipitation products (Al-OH, Al-Oxal, Al-Cit and Al-Tan) as well as Fe ones and 3) two pH values of 5.5 and 7.0. That is besides all possible interactions between the tested factors. The model of PZSE consists of three factors including; 1) two types of metal hydroxides (Al and Fe), 2) four forms of Al and Fe precipitation products and 3) four molar ratios (MR) of ligand/metal. Besides, all possible interactions between tested factors.
RESULTS AND DISCUSSION
Point of zero salt effect (PZSE) and the remaining charge (σp): Point of zero salt effect (PZSE) of non-modified and modified Al precipitation products formed in absence (Al-OH) or presence of organic ligands, oxalate (Al-Oxal), citrate (Al-Cit) or tannate (Al-Tan), at ligand/Al MR of 0.1 are graphically derived from Fig. 1 and listed in Table 1.
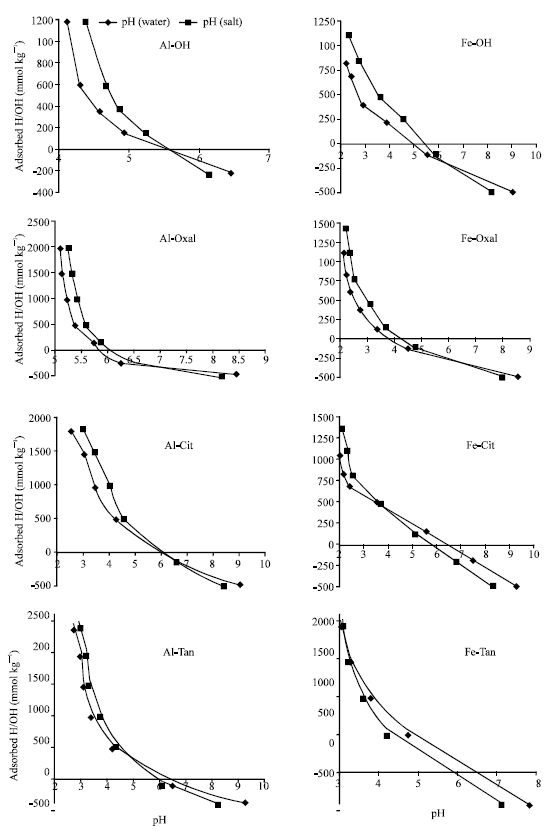 | |
| Fig. 1: | Point of zero charge (PZSE) and the remaining charge (σp) at the same pH, expressed as H+/OH¯ sorbed, for Al and Fe precipitates |
| Table 1: | Point of zero salt effect (PZSE) and cation exchange capacity (CEC, Cmolc kg-1) of Al and Fe precipitates formed in abscence or presence of organic ligands |
 | |
| *Molar ratio of ligand/metal | |
| Table 2: | Point of zero salt effect (PZSE) of Al and Fe precipitates formed at (Oxal or Tan)/Metal MR of 0.02 and 0.2 |
The results show that, co-precipitation of organic ligands with Al significantly (p = 0.001) shifted PZSE of the precipitation products to lower or higher pH values. In the presence of oxalate ligand, PZSE value (5.65) of non-modified form (Al-OH) shifted to higher pH value of 7.1. On the other hand, in the presence of citrate or tannate ligands, PZSE shifted to lower pH values of 5.45 and 4.85, for Al-Cit and Al-Tan, respectively. Also, the quantity and sign of the remaining charge (σp) of these precipitates are influenced by the co-precipitated ligand. The remaining positive charge of Al-OH increased in the presence of oxalate ligand, expressed as sorbed OH¯ (Fig. 1). On the other hand, in the presence of citrate or tannate ligands, σp of the precipitation products changed to be negative, expressed as sorbed H+. Increasing PZC value indicates decreasing surface acidity and vice versa (Mustafa et al., 2004). In agreement with the present results, Violante and Huang (1984) reported a decrease in PZC value of pseudoboehmites formed in the presence of citrate or tannate ligand. Decreasing PZSE of Al-Tan and Al-Cit, to lower pH values could be attributed to partial neutralization of positive charges through the replacement of H2O molecules coordinated with Al and proton dissociation of the organic functional groups (Liu and Huang, 1999). On the other hand, increasing PZSE of Al-Oxal to a higher pH value could be attributed to the high affinity and ability of oxalate to strongly complex Al (Reid et al., 1985; Fox and Comerford, 1990), which may retard proton dissociation of oxalate functional groups (-COOH). Besides, the low molecular weight and small size of oxalate ligand, promote its penetration in the precipitate, rather than surface precipitation, which may also retard proton dissociation. Also, increasing PZSE values could be attributed to alteration of surface properties as a result of ligand incorporation. Strauss et al. (1997) reported that, the PZC of goethite increased as surface area increased. Also, Zeltner and Anderson (1988) found an increase in PZSE of goethite from pH 7.0 to 8.8 with an increase in the specific surface area from 20 to 120 m2 g-1.
Unlike Al precipitates, co-precipitation of organic ligands with Fe markedly shifted PZSE of the precipitation products to lower pH values, Table 1. PZSE value (pH = 6.4) of non-modified Fe precipitate (Fe-OH) significantly (p = 0.001) decreased to be 6.1, 3.7 and 3.2 for Fe-Oxal, Fe-Cit and Fe-Tan, respectively. These results indicate that, co-precipitation of organic ligands with Fe leads to significant increase in surface acidity, particularly for tannate and citrate ligands. In agreement with these results, Yamaguchi et al. (1996) reported that, PZC of amorphous Fe hydroxide is 7.0, which is little higher than the value of the present study. Liu and Haung (1999) reported that, PZSE of iron oxide formed in the presence of citrate ligand decreased from 7.0 to 3.9 as the ligand concentration increased from 0.0 to 1 mM. The presence of citric acid during the formation of short-range ordered Fe oxide lead to substantial decrease in PZC value of the precipitation product (Xue and Huang, 1995). Regarding to the remaining charge (σp) of Fe precipitates, the results showed that, the remaining positive σp of Fe-OH increased in the presence of oxalate ligand (Fe-Oxal). On the other hand, in the presence of citrate (Fe-Cit) or tannate (Fe-Tan) ligand, σp reversed to be negative (Fig. 1). In agreement with these results, Liu and Huang (2001) found that, Fe oxide formed in the presence of citrate ligand (MR = 0.001) had lower PZSE and more negative charge at same pH.
Table 2 represents PZSE values of Al and Fe precipitates formed in the presence of oxalate or tannate ligands at two different MR (0.02 and 0.2). The results (Table 1 and 2) showed that, as the MR increased, PZSE of Al and Fe precipitates significantly (P ranged from 0.018 to 0.001) shifted to higher or lower pH values. As Oxal/Al MR increased from 0.0, 0.02, 0.1 to 0.2, PZSE of the precipitation products considerably shifted to higher pH values (from 5.65, 6.35, 7.1 to 7.9, respectively). Increasing PZSE values of Al-Oxal with MR could be attributed to proceed -OH and/or -OH2 replacement by oxalate ligand due to the increasing of the amount incorporated into the precipitate. On the other hand, as Oxal/Fe MR increased, PZSE values of Fe precipitates slightly shifted to lower pH values (from 6.4, 6.3, 6.1 to 5.95, respectively), which imply that, proton dissociation of oxalate functional groups (-COOH) coordinated to Fe is higher than those coordinated to Al. Increasing PZSE values of Al-Oxal versus decreasing those of Fe-Oxal could be attributed to differences in affinity between oxalate ligand and central Al and Fe (Violante and Huang, 1989: Kowing and Huang, 1979). In the presence of tannate ligand, as the MR increased, PZSE values markedly decreased from 5.65, 5.3, 4.85 to 3.6 and from 6.4, 3.9, 3.2 to 2.96 for Al-Tan and Fe-Tan, respectively. In agreement with these results, PZSE value (pH = 5.4) of aluminum oxide prepared in the presence of tannate ligand at MR of 0.01 (Violante and Huang, 1989, 1992) is close to the value (5.3) obtained for the present study at MR of 0.02. These results indicated that, co-precipitation of organic ligands with Fe leads to marked decreases in PZSE values, i.e., increasing surface acidity of Fe precipitates comparing with Al ones.
Cation exchange capacity (CEC): Cation exchange capacity (CEC), pH 5.5, of Al and Fe precipitates varied widely between the precipitates and ranged from 32.9 to 62.4 and 58.9 to 68 cmolc kg-1, respectively, Table 1. The corresponding values at pH 7.0 are generally higher (102 to 110%) and ranged from 33.6 to 68.5 and 62.4 to 70 mmol kg-1, respectively. These results indicated that, co-precipitation of organic ligands with Al or Fe significantly (p=0.001) enhanced CEC values of the precipitation products. As the molecular weight (MW) of organic ligand increased (oxalate < citrate < tannate), CEC values significantly (p=0.001) increased. Co-precipitation of organic ligands with Al lead to considerable increases in CEC values of the precipitation products (ranging from 150 to 200%) comparing with non-modified Al-OH, whereas, their co-precipitation with Fe lead to little increases (ranging from 107 to 114%) comparing with Fe-OH. The data (Table 1), also, showed that, CEC values of Fe precipitates are generally higher, but with different order of magnitude, than their correspondings of Al ones. CEC values (pH 5.5 and 7.0) of Fe-OH are much higher, on average 180%, than those of their corresponding of Al-OH. However, those of Fe-Oxal and Fe-Cit, are little higher (on average, 132 and 116%, respectively) than those of their correspondings of Al-Oxal and Al-Cit. On the other hand, those of Fe-Tan are slightly higher than (on average 108%) those of Al-Tan. The wide variation (180%) in CEC values between Fe-OH and Al-OH (the non-modified forms), could be attributed to the initial differences between the reactivity, i.e., proton dissociation, of surface inorganic (-OH and -OH2) groups as coordinated with Fe or Al. Besides, the difference in particle size between Fe and Al precipitates, in which Fe (hydr)oxide particles usually had lower size than Al ones.
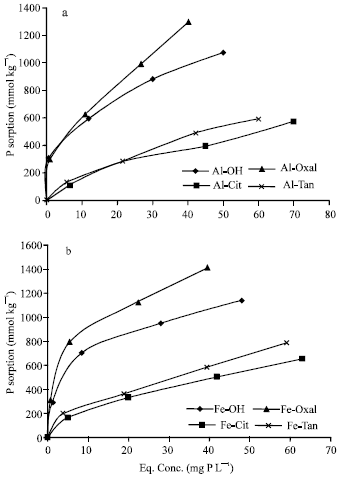 | |
| Fig. 2: | Isotherms of P sorption on Al (a) and Fe (b) precipitation products, at pH 5.5 |
On the other hand, the narrow variation between CEC values of Fe-Tan and Al-Tan, indicated that, the surface properties are greatly influenced by organic functional groups (-COOH and -OH) of tannate ligand. This could be attributed to the ability of tannate to cover a large area of surface precipitate due to its high MW (1701). i.e., the influence of inorganic functional groups (-OH or -OH2 coordinated with Al or Fe), on surface charge decreased as the MW of the co-precipitated ligand increased. In comparison with other reactive materials, CEC values of Fe and Al precipitates formed in the presence of organic ligands (in particular, tannate or citrate ones) are higher than those (48.3 and 58.1 cmolc kg-1) reported by Saha et al. (2002) for hydroxyaluminum- and hydroxyaluminosilicate-montmorillonite complexes at pH 7.0, respectively. These results verify the high reactivity of amorphous Al and Fe (hydr)oxides modified by organic ligands comparing with other reactive materials such as secondary clay minerals present in soil or sediments.
P sorption on Al and Fe precipitation products: Sorption isotherms of P on Al and Fe precipitation products, at pH 5.5 are shown in Fig. 2. The results show the high capacity of both Al and Fe precipitates to sorb soluble P. The total quantities (Fig. 2a) of sorbed P varied significantly (p = 0.001) between Al precipitates and ranged from 109 to 1300 mmol kg-1. The isotherms show the high potential ability of Al-Oxal and Al-OH to sorb additional quantities of P, whereas Al-Cit and Al-Tan had limited ability in this connection. These results proved that, co-precipitation of oxalate ligand with Al enhanced the capacity of the precipitates to sorb P, whereas co-precipitation of citrate or tannate ligands leads to considerable decreases in P sorption capacity. In agreement with these results, the quantity of P sorbed on aluminum hydroxide precipitates formed in the presence of tannate ligand (572 mmol kg-1, at pH 5.0) as reported by Violante and Pigna (2002), is close to that (590.3 mmol kg-1, at pH 5.5) sorbed on Al-Tan of the present study. Violante and Huang (1989) reported that, Al precipitation products formed in the presence of strong chelating agents (Cit, Tart or Tan) sorbed less P than those formed in the presence of aspartate or Cl¯ ligands. Due to its high molecular weight (MW = 1701), tannate ligand should cover a large area of the surface of aluminum precipitates, masking many P sorption sites (Kwong and Huang, 1981).
Regarding to sorption isotherms of P on Fe precipitates at pH 5.5, Fig. 2b shows that, the quantities of P sorbed on Fe precipitates (164 to 1413 mmol kg-1) are significantly higher (p=0.01) than their correspondings of Al precipitates. Similar to Al precipitates, co-precipitation of oxalate ligand with Fe efficiently increased the capacity of the precipitation products to sorb P, whereas, co-precipitation of citrate or tannate ligands suppressed P sorption capacity. Both Fe-Oxal and Fe-OH had high potential ability to sorb additional quantities of P, whereas Fe-Cit and Fe-Tan had limited potential ability in this connection. The quantity of P sorbed on amorphous Fe hydroxide (Fe-OH) of the present study is lower than that (1761 mmol kg-1) sorbed on hydrous ferric oxide gel at pH 5.5 as reported by Ryden et al. (1987). However, they used P concentration (2.5 mmol L-1), higher than that of the present study (2.25 mmol L-1).
Sorption isotherms of P on Al and Fe precipitates at pH 7.0 are shown in Fig. 3. The results show that, as pH increased from 5.5 to 7.0, P sorption capacity of all Al and Fe precipitates significantly (p=0.001) decreased. Total quantities of P sorbed on Al precipitates at pH 7.0 (75 to 1182 mmol kg-1) are markedly lower than (representing 68 to 91%) those sorbed at pH 5.5. Also, the quantities of P sorbed on Fe precipitates at pH 7.0 (126 to 1020 mmol kg-1) are markedly lower (representing 72 to 77%) than those sorbed at pH 5.5.
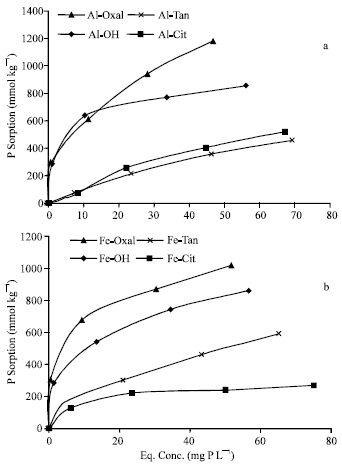 | |
| Fig. 3: | Isotherms of P sorption on Al (a) and Fe (b) precipitation products, at pH 7.0 |
Similar to the trends obtained at pH 5.5, Al-Oxal, Fe-Oxal and to a lesser degree, Al-OH and Fe-OH, had high capacity and potential ability to sorb P, whereas Al-Cit, Al-Tan, Fe-Cit and Fe-Tan, had low capacity and limited potential ability to sorb P.
Regarding to proton consumption with phosphate sorption on Al and Fe precipitates, the results show that, except for Al-OH, no protons were consumed with phosphate sorption on Al and Fe precipitates at pH 5.5. Small quantities of protons (5.2 to 25.8 mmol kg-1) were consumed with phosphate sorption on Al-OH. At pH 7.0, except for Fe-Tan, fixed amounts of protons (20.6 to 30.9 mmol kg-1) were consumed with phosphate sorption on Al and Fe precipitates. These quantities are independent of both, the quantity of P sorbed and the type of adsorbent. In case of Fe-Tan, higher quantities of proton (30.9 to 401.7 mmol kg-1) were consumed with phosphate sorption. Proton consumption increased as the phosphate sorption on Fe-Tan increased. Phosphate adsorption involves a ligand exchange reaction with OH2 and OH that were coordinated on the surface (Hingston et al., 1967, 1972).
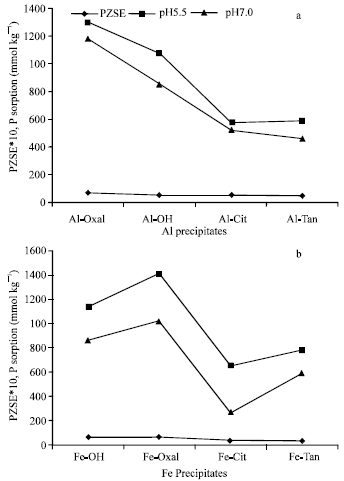 | |
| Fig. 4: | PZSE and P sorption on Al (a) and Fe (b) precipitates at pH 5.5 and 7.0 |
If no source affects the pH change, the determination of proton migration with adsorption indicates the extent of the ligand exchange reaction with OH groups. Protons, which migrated with phosphate dissociation, however, would interfere with this determination. Just for comparison between phosphate sorption behavior on various Al and Fe precipitates, the results suggested that; at pH 5.5, except for Al-OH, exchange between phosphate ion and OH2 group is the expected mechanism for phosphate adsorption on all Al and Fe precipitates, whereas, OH groups might be much more involved than OH2, in the exchange with phosphate adsorption on Al-OH. At pH 7.0, OH groups is much more expected to be involved in phosphate sorption on Al and Fe precipitates, particularly for Fe-Tan, on which the reaction seemed to be stoichiometric, since the ratio between H+ consumption and phosphate sorption is approaching 1: 1.
Surface properties and P sorption: Isotherms of P sorption showed that all Al and Fe precipitates had the ability to sorb considerable quantities of P at pH (7.0) higher than, or almost equal to (in case of Al-Oxal), their PZSE values. These results verified that specific adsorption is the expected mechanism for P retention, rather than both, nonspecific one, formation of outer-sphere complex and/or formation of separate phase. Variations in the capacity of modified and non-modified Al and Fe precipitates to sorb P could be discussed based on PZSE values. Figure 4a shows positive relationship between PZSE values of Al precipitates and the quantities of P sorbed at pH 5.5 and 7.0. As PZSE of Al precipitates decreased from 7.1 to 4.85, P sorbed at pH 5.5 and 7.0, markedly decreases from 1300 to 590 and 1182 to 460 mmol kg-1, respectively. Regarding to Fe precipitates, Fig. 4b shows positive relationship between PZSE values and the quantity of P sorbed on Fe precipitates. In despite of some deviations showed in Fig. 4b, the general trend showed considerable decreases in the quantities of sorbed P at pH 5.5 and 7.0. These decreases are synonymous to decreases in PZSE values of the precipitates. Also, the capacity of Al and Fe precipitates to sorb P could be discussed with the remaining charge (σp) at PZSE. The precipitates of Al-Oxal, Al-OH, Fe-Oxal and Fe-OH, which had positive σp, had much higher capacity to sorb P than those (Al-Cit, Al-Tan, Fe-Cit and Fe-Tan) had negative σp. The remaining positive charge (σp) refers to that, anion sorption is much more preferable than cations and vice versa.
P-desorption: The quantities (29 to 645 mmol kg-1) of P desorbed from Al precipitates varied significantly (p = 0.001) between the precipitates and represented 22 to 60% of those previously sorbed at pH 5.5 (Fig. 5a). The desorbed quantities increased with sorption density. Generally, P desorption showed two different trends. The first one observed at relatively low sorbed quantities (roughly, less than 600 mmol P kg-1), on which the desorbed quantities were relatively low and represented 22 to 31% of the previously sorbed. The second one observed at higher sorbed P, on which the desorbed quantities increased and represented 45 and 60% of the previously sorbed on Al-Oxal and Al-OH (the two precipitates had high sorbed quantities), respectively. Similar trends were obtained for P desorbed from the previously sorbed on Al precipitates at pH 7.0 (Fig. 5b). The desorbed quantities varied significantly (P = 0.001) between the precipitates and represented 20 to 62% of the previously sorbed ones, which are identical to those obtained at pH 5.5. However, the quantities of P sorbed at pH 7.0 are lower than those at pH 5.5, indicating the ability of Al precipitates to sequester sorbed P decreased as pH increased. At low sorbed P, the quantities of P desorbed represented 20 to 35% of the previously sorbed.
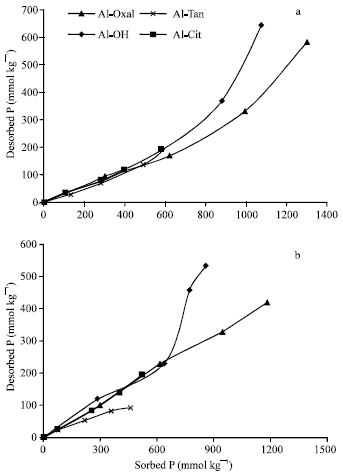 | |
| Fig. 5: | Desorption of P as a function of the previously sorbed on Al precipitation products, at pH 5.5 (a) and 7.0 (b) |
At higher sorbed P, the quantities desorbed from Al-OH increased and represented 60%, whereas, those of Al-Oxal remain stable at 35%. These results verify that, at low sorbed P the ability of all Al precipitates to sequester sorbed P are relatively high. As sorbed P increased, the ability markedly decreased for Al-OH and Al-Oxal. The higher ability of Al-Oxal to retain sorbed P, comparing with Al-OH, could be attributed to that, PZSE value and remaining positive charge (σp) of Al-Oxal are higher than those of Al-OH, which indicate its high affinity to anions.
The quantities of P desorbed from Fe precipitates represented 24 to 70% and 31 to 85% of those previously sorbed at pH 5.5 and 7.0, respectively, (Fig. 6a and b). The results showed that, Fe-Tan had the lowest ability to sequester sorbed P, since the desorbed quantities represented 51 to 70% and 51 to 85% of the previously sorbed at pH 5.5 and 7.0. That is besides, its low capacity to sorb P. Regarding to the other three precipitates (Al-OH, Al-Cit and Al-Oxal), at low sorbed P, the desorbed quantities represented only 21 to 32% and approximately, 37%, of the previously sorbed at pH 5.5 and 7.0, respectively.
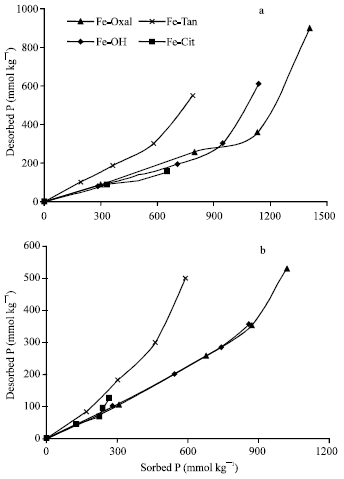 | |
| Fig. 6: | Desorption of P as a function of the previously sorbed on Fe precipitation products, at pH 5.5 (a) and 7.0 (b) |
At higher sorbed P, the desorbed quantities increased to be 55 and 40% of the previously sorbed on Al-OH at pH 5.5 and 7.0, respectively. Their correspondings of Al-Oxal are 64 and 50%, respectively. However, the quantities of P sorbed at pH 5.5 are much higher than those sorbed at pH 7.0, which could be a reason for the increments showed in percentages of P desorbed at pH 5.5. The higher reversibility of P sorbed on Fe-Tan comparing with the other three precipitates could be attributed to that, Fe-Tan had the lowest PZSE value and the highest positive remaining charge (σp). This trend was not clear with Fe-Cit that had negative σp due to the relatively low quantities of sorbed P.
In general, increasing P reversibility with sorption density, indicating that, more than one kind of sites is responsible for P retention. The first one had high affinity for P, on which formation of inner sphere complex is the mechanism expected to be responsible for P retention. The first kind of sites is more probably responsible for the low and constant P reversibility (21 to 35%) obtained for all Al and Fe precipitates at low sorption density and at wide range of pH (from 5.5 to 7.0). The second one had low affinity for P, on which, formation of outer sphere complex, i.e., nonspecific adsorption, is the expected mechanism.
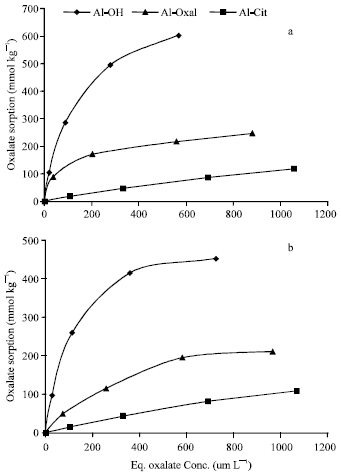 | |
| Fig. 7: | Isotherms of oxalate sorption on Al precipitation products at pH 5.5 (a) and 7.0 (b) |
This kind of sites may responsible for higher P reversibility recorded for higher P sorption capacity. In agreement with these findings, models of ion sorption by oxides, developed by Benjamin and Leckie (1981); Hiemstra et al. (1989 a, b), suggested that the surface of oxides is composed of multiple sorption sites with different binding strengths.
Oxalate sorption: Isotherms of oxalate sorption on Al precipitates at pH 5.5 and 7.0 are shown in Fig. 7. The quantities of oxalate sorbed on Al precipitates varied widely between the precipitates and ranged from 18 to 605 and 15 to 453 mmol kg-1, at pH 5.5 and 7.0, respectively. The quantities of oxalate sorbed at pH 7.0 are lower than (representing 75 to 91% of) those sorbed at pH 5.5. These results showed that, co-precipitation of oxalate or citrate ligands with Al suppress the capacity of the precipitation products to sorb oxalate ions. The quantity of oxalate sorbed on Al-OH at pH 5.5 could be compared with that (523 mmol kg-1) sorbed on synthetic aluminum hydroxysulphate complex, at pH 5 and equilibrium concentration of oxalate of 1 mmol L-1, as reported by (Violante et al., 1996).
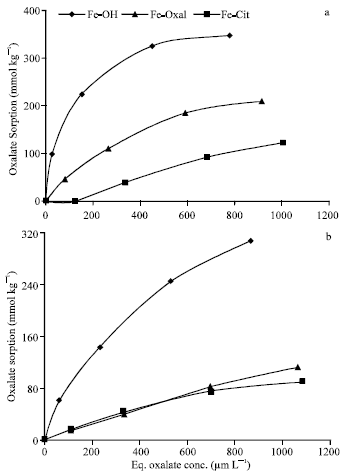 | |
| Fig. 8: | Isotherms of oxalate sorption on Fe precipitation products at pH 5.5 (a) and 7.0 (b) |
Figure 8 shows the isotherms of oxalate sorption on Fe precipitates at pH 5.5 and 7.0. The quantities of oxalate sorbed on Fe precipitates at pH 5.5 (0 to 348 mmol kg-1) are much higher than those sorbed at pH 7.0 (15 to 307 mmol kg-1). Similar to Al precipitates, the non-modified form (Fe-OH) had the highest capacity to sorb oxalate ions at pH 5.5 and 7.0. i.e., Co-precipitation of citrate or oxalate ligands with Fe suppress the capacity of the precipitation products to sorb oxalate ions. In general, the capacity of Fe precipitates to sorb oxalate ions are markedly lower and represented approximately 57% of those of Al precipitates. The capacity of Al and Fe precipitates to sorb oxalate could be discussed with PZSE values. As PZSE values of Fe precipitates decreased from 6.4, 6.1 to 3.7 for Al-OH, Fe-Oxal and Fe-Cit, respectively, the quantities of oxalate sorbed decreased from 348, 210 to 122 (pH 5.5) and from 307, 112 to 90 mmol kg-1 (pH 7.0), respectively. Regarding to Al precipitates, as PZSE decreased from 5.65 to 5.45 for Al-OH and Al-Cit, respectively, the quantities of oxalate sorbed decreased from 605 to 118 mmol kg-1 (pH 5.5) and from 453 to 107 mmol kg-1 (pH 7.0), respectively. (Data of Al-Oxal showed deviation from this trend.) The quantities of oxalate sorbed on Al-Tan and Fe-Tan were relatively low and it was difficult to be analyzed, particularly at lower concentrations, so it suggested to be excluded.
In conclusion, the presence of organic ligands during the formation of Al and Fe hydroxides significantly altered the surface properties of the precipitation products, point of zero charge (PZSE) and the remaining charge (σp) at the same pH, as well as cation exchange capacity (CEC). These properties significantly varied as the co-precipitated ligand and metal hydroxide varied, which lead to significant differences on sorption/desorption characteristics of these precipitates pertaining to inorganic (phosphate) and organic (oxalate) ligands. Based on their high capacity to sorb and sequester sorbed inorganic and organic ligands, these materials could be efficiently used in pollution abatement; decontamination of polluted water and chemi-immobilization of pollutants in soil. However, sorption/desorption characteristics of heavy metals on these materials need to be explored. Also, these precipitates merit substantial research concerning their stability and phase transformation from amorphous to crystalline form under different soil conditions.
REFERENCES
- Bolan, N.S., R. Naidu, J.K. Syers and R.W. Tillman, 1999. Surface charge and solute interactions in soils. Adv. Agron., 67: 87-140.
Direct Link - Fox, T.R. and N.B. Comerford, 1990. Low-molecular-weight organic acids in selected forest soils of the Southeastern USA. Soil Sci. Soc. Am. J., 54: 1139-1144.
CrossRefDirect Link - Goldberg, S., I. Lebron, D.L. Suarez and Z.R. Henidi, 2001. Surface characterization of amorphous aluminum oxides. Soil Sci. Soc. Am. J., 65: 78-86.
Direct Link - Hingston, F.J., R.J. Atkinson, A.M. Posner and J.P. Quirk, 1967. Specific adsorption of anions. Nature, 215: 1459-1461.
CrossRef - Hingston, F.J., A.M. Posner and J.P. Quirk, 1972. Anion adsorption by goethite and gibbsite. I. The role of the proton in determining adsorption envelopes. J. Soil Sci., 23: 177-192.
CrossRefDirect Link - Jara, A.A., S. Goldberg and M.L. Mora, 2005. Studies of the surface charge of amorphous aluminosilicates using surface complexation models. J. Colloid Interface Sci., 292: 160-170.
PubMedDirect Link - Kinniburgh, D.J., J.K. Syers and M.L. Jackson, 1975. Specific adsorption of trace amounts of calcium and strontium by hydrous oxides of iron and aluminum. Soil Sci. Soc. Am. Proc., 39: 464-470.
Direct Link - Liu, C. and P.M. Huang, 1999. Atomic force microscopy and surface characteristics of iron oxides formed in citrate solutions. Soil Sci. Soc. Am. J., 63: 65-72.
Direct Link - Murphy, J. and J.P. Riley, 1962. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta, 27: 31-36.
CrossRefDirect Link - Ryden, J.C., J.K. Syers and R.W. Tillman, 1987. Inorganic anion sorption and interactions with phosphate sorption by hydrous ferric oxide gel. J. Soil Sci., 38: 211-217.
Direct Link - Sakurai, K., Y. Ohdate and K. Kyuma, 1988. Comparison of salt titration and potentiometeric titration methods for the determination of zero point of charge (ZPC). Soil Sci. Plant Nutr., 34: 171-182.
Direct Link - Strauss, R., G.W. Brummer and N.J. Barrow, 1997. Effects of crystallinity of goethite preparation and properties of goethite of differing crystallinity. Eur. J. Soil. Sci., 48: 87-99.
Direct Link - Uehara, G. and G.P. Gillman, 1980. Charge characteristic of soils with variable and permanent charge minerals: I. Theory. Soil Sci. Soc. Am. J., 44: 250-252.
Direct Link - Violante, A. and P.M. Huang, 1989. Influence of oxidation treatments on surface properties and reactivities of short-range ordered precipitation products of aluminum. Soil Sci. Soc. Am. J., 53: 1402-1407.
Direct Link - Violante, A., M.A. Rao, A. De chiara and L. Gianfreda, 1996. Sorption of phosphate and oxalate by a synthetic aluminium hydroxysulphate complex. Eur. J. Soil Sci., 47: 241-247.
CrossRefDirect Link - Violante, A. and L. Gianfreda, 1995. Adsorption of Phosphate on Variable Charge Minerals: Competitive Effect of Organic Ligands. In: Environmental Impact of Soil Component Interactions, Huang, P.M., J. Berthelin, J.M. Bollag, W.B. McGill and A.L. Page (Eds.). CRC Lewish Publications, Boca Raton, pp: 27-36.
- Gillman, G.P., 1979. A proposed method for the measurement of exchange properties of highly weathered soil. Aust. J. Soil Res., 17: 129-139.
Direct Link