
Saad Osman Abdalla
Faculty of Applied Sciences, Sohar University, Sohar, Sultanate of Oman
Safaai Deris
Faculty of Computer Science and Information Systems, University of Technology Malaysia, UTM Skudai, Johor, Malaysia
Information Technology Journal
Year: 2005 | Volume: 4 | Issue: 2 | Page No.: 189-196







ABSTRACT
Novel researchers in the area of protein secondary structure prediction using artificial neural networks take a considerable time to get the most important knowledge in this area. This study was conducted to make the most foundational and directive knowledge in protein biological aspects, protein secondary structure prediction and protein neural network predictors get elucidated. Several neural network methods have contributed and influenced significantly the field of bioinformatics in general and the area of secondary structure prediction from protein sequences in specific. Present research suggest that there is a lot of work to be done to fully exploit artificial neural networks in this area.
PDF Abstract XML References Citation
How to cite this article
Saad Osman Abdalla and Safaai Deris, 2005. Predicting Protein Secondary Structure Using Artificial Neural Networks: Current Status and Future Directions. Information Technology Journal, 4: 189-196.
DOI: 10.3923/itj.2005.189.196
URL: https://scialert.net/abstract/?doi=itj.2005.189.196
DOI: 10.3923/itj.2005.189.196
URL: https://scialert.net/abstract/?doi=itj.2005.189.196
INTRODUCTION
Protein is considered as series of amino acids linked together into contiguous chains. The production of proteins in a cell is governed by codes and information transferred to the DNA and RNA in the organism’s cell. The DNA of an organism encodes its proteins in a sequence of nucleotides, namely: adenine, cytosine, guanine and thymine. These nucleotides considered as information that governs the process of protein synthesis[1].
The amino acids consist of a carbon as a central atom linked to hydrogen or oxygen which forms molecules that connect with each other.
There are 64 different amino acids correspond from four nucleotides that make the universal genetic code (Table 1) but only twenty different types of amino acids work as basic building units of a protein (Table 2)[2,3].
The amino acid sequence is the primary structure of a protein. It is usually represented by the one letter notation of the amino acids. Amino acids combine to form a protein through polypeptide bonds and here the protein could be considered as:
| Table 1: | The universal genetic code |
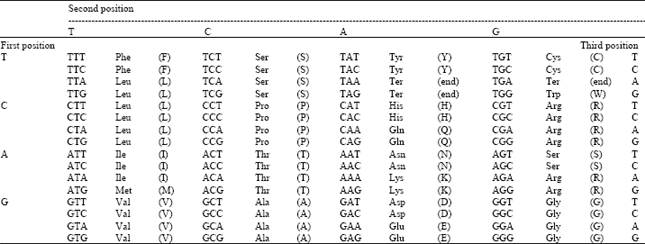 | |
polypeptide chain and the amino acids as residues. Anyhow the sequence direction is very important and usually represented from the amino (N) terminus to the carboxyl (C) terminus[1-3].
PROTEIN SEQUENCES AND STRUCTURES
The sequence of amino acids in a protein chain forms the protein structure. Protein structures could be classified into four levels or classes: primary, secondary, tertiary and quaternary structure[1-3]. When the sequences of primary structures tend to arrange themselves into regular formations, these units are referred to as secondary structure.
The angles and hydrogen bond patterns between the backbone atoms are determinant factors in protein secondary structure. Moreover, secondary structure is subdivided into three parts: α-helix, β-sheet and loop[4]. α-helices and β-sheets are the most common form of secondary structure, in proteins. Loops usually serve as connection points between α-helices and β-sheets and they do not have even patterns like α-helices and β-sheets. However, in most cases any patterns which are not α-helices or β-sheets are considered as loops[1,3].
Different folds that often possess similar arrangements of a two to four consecutive recurring units of secondary structures are called super-secondary structures or motifs[5-7].
The three-dimensional structure of the protein, which is formed from the secondary structures as subunits elements, is known as the protein’s tertiary structure. Forces like hydrophobic side-chains in the core of proteins, hydrogen bonds, van der Waals forces and oppositely charged amino acid side-chains are considered as the driving force of tertiary structure formation[8,9]. An individual protein that its independent fold or substructures form a three dimensional structure of the protein is known as quaternary structure. This is true for some proteins because they can not work in isolation; haemoglobin and RNA polymerase are examples of such proteins[1,3]. However, the most important obstacle in protein folding problem is that although there is considerable amount of information about the forming a three dimensional structure of a protein from simple sequences, protein folding fails in vitro as it does not do in vivo[10,11].
In the mid of nineties, bacterial genome was the first to be sequenced and at the year 2001, the first draft of human genome has been announced. However, until now there are over 65 organisms have been entirely sequenced[12-15].
| Table 2: | The twenty types of amino acids that forms the proteins |
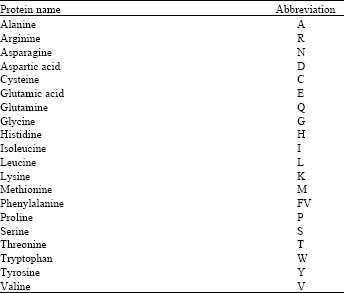 | |
METHODS OF DETERMINING PROTEIN STRUCTURE
Three-dimensional structures of a protein can be determined in very details that describe the relative position of a single atom within the protein using two laboratory methods: x-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. X-ray crystallography is lengthy and complicated process. It requires high level of technical ability in the laboratory to reach to an inference of the x-ray diffraction patterns. In NMR spectroscopy the protein is put in a strong magnetic field and subjected to Radio Frequency (RF) pulses. This will force the protein to emit RF radiation. Then information of protein structure can be inferred from the frequencies and intensities of the emitted radiation. NMR process is not easy and there are many biochemical constraints in this process[16]. However, these methods require several months or even years of laboratory work and they are not viable for some proteins. This led to the fact that introducing procedures or processes of protein sequence prediction could save a considerable time and efforts[16-20].
Proteins of the same family are known as homologous proteins or homologs. Proteins change conservatively through evolution and similar proteins express similar functions[21]. Comparing two different proteins homologs is known as protein sequence alignment. One of three states occurs in the alignment process: substitution which is the replacement of one or more residues, deletion which the removal of one or more residues, insertion which is the addition of one or more residues[22]. Sequence alignment is performed when different protein sequences are put in rows while columns represent regions of mach or mismatch. When aligning two sequences, regions of mismatch in the other sequence are deleted and represented by dashes. These deleted regions are called gaps.
Alignments that contain two protein sequences are known as pairwise alignment, while those contain many sequences are known as multiple alignments. Researchers showed that similar protein sequences usually reflect similar functions, although there are exceptions of the previous conclusion[23,24].
Since Anfinsen[25] concluded that the amino acid sequence is the only source of information to survive the denaturing process and hence the structured information must be somehow specified by the primary protein sequence, researchers have been trying to predict secondary structure from protein sequence. Anfinsen’s hypothesis suggests that an ideal theoretical model of predicting protein secondary structure form its sequence should exist anyhow.
The gap between known structures and known sequences is growing wider. It is known that protein structure is difficult to be predicted from the protein sequence However, considerable works has been done in this area[11,26]. The present research showed that membrane helices can be predicted much more accurately than globular helices and internal helices are predicted less accurately[27-29].
Using small datasets of protein in experimental methods to predict secondary structure adversely affected the accuracy of methods[30-32]. At present there is enough data for experimental methods to boost their accuracy[33]. Many algorithms and methods have been applied to the secondary structure of protein. Most known methods are: Statistical Methods[34,35]. Nearest-neighbour algorithms[36,37] and Neural networks methods[38,39]. Although many workers on these methods claimed accuracy as high as 78 %, using correct data set made the range of accuracy drop to the level of 60%[40,41].
The reliability index provides a good tool to study some key regions predicted at high levels of expected accuracy. Accuracy of prediction is correlated with the reliability index. This means that residues with higher reliability index are predicted with higher accuracy than others[40]. However, alignment is key point here, because bad alignments lead to bad prediction.
However, it is not always true to combine different prediction methods to reach higher accuracy. For some methods like EVA, combining different methods decreased accuracy over the best individual methods, although averaging over the better ones was better than averaging the best ones[42-44].
Protein secondary structure formation is influenced by long-range interactions and the environment[45]. Consequently, stretches of adjacent residues can be found in different secondary structure states this non-local effects are contained in the exchange patterns of protein families[46,47].
PREDICTION USING NEURAL NETWORKS
As an efficient pattern classification tool[48], neural network has been used in protein structure prediction problem by many researchers[28,41,49]. Qian and Sejnowski[55] followed by Bohr et al.[57] greatly influenced the approach of predicting protein structure by their work when first introduced neural networks in this area. Artificial neural networks combined with evolutionary information showed the capability and the potential of the system and paved the way of using this powerful tool in the bioinformatics field[56,59]. The high degree of prediction of protein secondary structure showed that the performance of neural networks exceeded the performance of other systems[49,50,58]. Nevertheless, neural networks has been extended to predict the location of active sites and content of protein[51,52].
Using an advanced neural network design, Baldi et al.[53] have improved the level of accuracy in predicting β strand pairings. Their bidirectional recurrent neural network outperformed the work of many researchers.
NEURAL NETWORKS ARCHITECTURES
Various architectures of artificial neural networks had been described by many researchers[48,54,55]. In this section we will explore some of the foundational architectures that had been used in protein secondary structure prediction. Inside the neural network (Fig. 1), many types of computational units exist; the most common type sums its inputs (xi) and passes the result through a nonlinear approximation or activation function (Fig. 2) to yield an output (yi). Thus the output (yi) = fi (xi), where fi is the transfer function of each unit. This is summarized in Eq. 1 and 2.
| (1) |
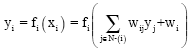 | (2) |
where, wi is the bias or threshold of the unit i.
A transfer function may be a linear function like the identity function of the regression analysis and hence the unit i is a linear unit. However, in Artificial Neural Networks most of the time the transfer functions are non linear like sigmoid and threshold logic functions.
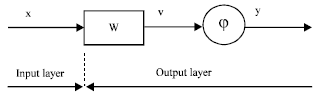 | |
| Fig. 1: | Basic graphical representation of a single neuron Artificial Neural Networks |
Bounded activation functions are often known as squashing functions. When f is a threshold or bias function, then:
| (3) |
Then Eq. 4 shows a sigmoid transfer function of type logistic transfer function which can estimate the probability of binary event.
| (4) |
One of the most important properties of Artificial Neural Networks is that they can approximate any reasonable function to any degree of precision[60,61]. If we have a continues function y = f(x) where, both y and x are one dimensional units and if x changes in the interval [0, 1], thus the value of x within a precision ε, where f is continuous over the compact interval [0, 1], there exists an integer n such that:
| (5) |
Then f can be approximated with a the function g(x) = f(k/n) for any x in the interval [(k-1/n, k/n)] and any unit representing k = 1,…,n.
If the data of our Artificial Neural Networks is assumed to be consisting of a set of independent input-output pairs Di = (di , ti) where, di is the input for unit i and ti is the output for unit i. The Artificial Neural Networks operation is a deterministic one in as seen in Eq. 6.
| (6) |
Hence inputs d could be assumed as independent of the parameter w, using the Bayesian inference, Eq. 6 can be transformed into[72]:
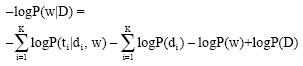 | (7) |
In the case of Gaussian regression, the probabilistic model assuming that the covariance matrix is diagonal and that there are n output units indexed by j, then:
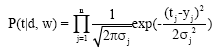 | (8) |
The derivative of the log likelihood E with respect to an output yi is shown in Eq. 9 which really the regular Least Mean Square (LMS) error function.
| (9) |
For a Artificial Neural Networks that classify an input into two classes (a and a¯), the target output can represented as 0 or 1. This model is a binomial model and can be estimated by a sigmoidal transfer function as shown in Eq. 10.
| (10) |
The relative entropy between the output distribution and the observed distribution is expressed by:
| (11) |
If the output transfer function is the logistic function, then:
| (12) |
| (13) |
Consequencely, in binomial classification, the output transfer function is logistic and the likelihood error function is the relative entropy between the predict distribution and the target distribution.
If the classification task of our Artificial Neural Networks has n possible classes (a1,…, an) for a given input d, then target out put t is a vector with a single 1 and n-1 zeros. However, Eq. 14-17 summarises the multi classes classification of Artificial Neural Networks.
| (14) |
| (15) |
| (16) |
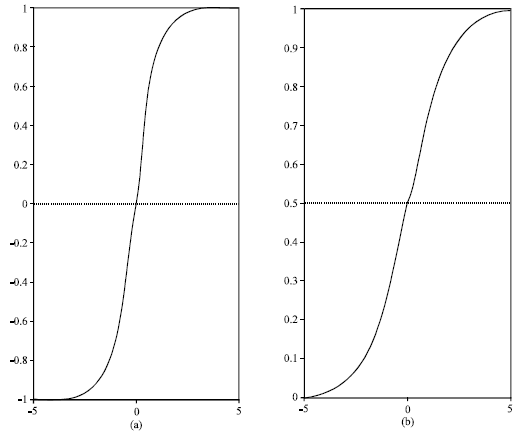 | |
| Fig. 2: | The sigmoid function conventionally used in feedforward Artificial Neural Networks a. sigmoid unipolar and its derivative function. b. sigmoid bipolar and its derivative function |
| (17) |
The main differences among these Artificial Neural Networks exist in topology where connectivity of nodes, methods of training and applications of the network differ.
One of the frequently used Artificial Neural Networks is the feedforward Artificial Neural Networks trained with backpropagation for rule extraction purposes. It is termed feedforward because information is provided as input and propagated in a forward manner, with each computational unit (perceptron) integrating its inputs and firing according to its specific non-linearity.
The simplest way to reduce the network error is by changing the connections according to the derivative of the error with respect to the connections, in a process known as the gradient descent. This is often referred to as back-propagating the error through the neural network[62,63]. To avoid being trapped in local minima, in practise, the actual training is typically performed by a variant of this algorithm that permits up-hill of the curve moves[59,64].
With enough hidden units neural networks can learn to separate any set of patterns. Typical applications require to extract particular features (underlying rules) present in the patterns rather than to learn the known examples. A successful extraction of such features permits the network to generalise, i.e., to also correctly classify patterns that have not been learned explicitly. Generalisation requires a balance between the number of training examples (enough to enable feature extraction) and the number of connections (enough to separate patterns). As a rule-of-thumb the number of connections should be an order of magnitude lower than the number of patterns to avoid over-fitting the training data (this learning exactly of the training set is also referred to as over-training)[59,64].
CONCLUSIONS
Artificial Neural Networks proved that they have the ability to making complex decisions based on the unbiased selection of the most important factors from a large number of competing variables. This is particularly important in the area of protein structure determination, where the principles governing protein folding are complex and not yet well understood.
Since the neural network can be trained to map specific input signals or patterns to a desired output, information from the central amino acid of each input value is modified by a weighting factor, grouped together then sent to a second level of a hidden layer where the signal is clustered into an appropriate class, the have great opportunities in the prediction of proteins secondary structures.
However, perdition of protein secondary structure can not be completely accurate due to the facts that the assignment of secondary structure may vary up to 12% between different crystals of the same protein. In addition, â-strand formation is more dependent on long-range interactions than β-helices and there should be a general tendency towards a lower prediction accuracy of α-strands than β-helices. However, this suggests that there still room of improvement since the accuracy of 88% has not reached yet.
ACKNOWLEDGMENTS
We would like to thank the late Dr. Nassrudin Zenon, Faculty of Computer Science and Information Systems, Universiti Teknolgi Malaysia, for valuable recommendations in this topic. We would also like to thank Professor Joachim Diederich, Professor of Computer Science, University of Queensland for seminars and discussion in this topic and Support Vector Machines topics.
REFERENCES
- Wright, P.E. and H.J. Dyson, 1999. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol., 293: 321-331.
PubMed - Rost, B., C. Sander and R. Schneider, 1994. PHD-an automatic server for protein secondary structure prediction. CABIOS, 10: 53-60.
PubMed - Koh, I.Y., V.A. Eyrich, M.A. Marti-Renom, D. Przybylski and M.S. Madhusudhan et al., 2003. EVA: Evaluation of protein structure prediction servers. Nucleic Acids Res., 31: 3311-3315.
PubMed - Liu, J. and B. Rost, 2003. NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res., 31: 3833-3835.
CrossRef - Qian, N. and T.J. Sejnowski, 1988. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol., 202: 865-884.
CrossRefPubMedDirect Link - Pollastri, G., D. Przybylski, B. Rost and P. Baldi, 2002. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins, 47: 228-235.
PubMed - Holley, H.L. and M. Karplus, 1989. Protein secondary structure prediction with a neural network. Proc. Natl. Acad. Sci. USA., 86: 152-156.
Direct Link - Rost, B. and C. Sander, 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol., 232: 584-599.
PubMed - Rost, B. and C. Sander, 1992. Exercising multi-layered networks on protein secondary structure. Int. J. Neural Syst., 3: 209-220.
Direct Link