
Ales Eichmeier
Mendel University, Faculty of Horticulture, Mendeleum Institute of Genetics and Plant Breeding, Valticka 334, 691 44, Lednice, Czech Republic
Petr Kominek
Crop Research Institute, Drnovsk� 507, 161 06 Prague 6-Ruzyn, Czech Republic
International Journal of Virology
Year: 2014 | Volume: 10 | Issue: 4 | Page No.: 263-271







ABSTRACT
A Real-Time RT-PCR (Reverse Transcription-Polymerase Chain Reaction) TaqMan® assay was used for the specific detection and absolute quantification of the Grapevine fanleaf virus (GFLV) in the infected tissues. The assay was targeted to a conserved region located in the 2BMP (movement protein) gene of the GFLV RNA2 molecule. This method was successfully used for the detection and quantification of GFLV in infected grapevines from South Moravia which were evaluated three times in 2011 during major phenophases. Moreover, DAS-ELISA detection was applied for the same samples to verify the higher sensitivity and reliability of Real-Time RT-PCR TaqMan®. The end-point dilution of ELISA was determined to calculate the detection limit of this method for GFLV. The Real-Time RT-PCR TaqMan® assay detected GFLV in all eight of the Moravian GFLV isolates tested, while DAS-ELISA was unable to detect GFLV in four of them. Isolate HV5 was easily detected by ELISA in 2004, when it was collected in the field. In the current experiment, after several years of cultivation in a screenhouse, the concentration of GFLV decreased under the detection limit of ELISA. The detection limit, calculated as the amount of viral RNA2 molecules present in a tested sample, for GFLV detection using Real-Time RT-PCR TaqMan® was approximately 1000-fold higher than that of ELISA which makes the method suitable for certification of grapevine propagation materials. GFLV detection was possible all the year, using phloem tissues.
PDF Abstract XML References Citation
Received: August 27, 2014;
Accepted: November 12, 2014;
Published: December 01, 2014
How to cite this article
Ales Eichmeier and Petr Kominek, 2014. Detection of Moravian Isolates of GFLV: Comparison of Real-Time RT-PCR and
ELISA. International Journal of Virology, 10: 263-271.
DOI: 10.3923/ijv.2014.263.271
URL: https://scialert.net/abstract/?doi=ijv.2014.263.271
DOI: 10.3923/ijv.2014.263.271
URL: https://scialert.net/abstract/?doi=ijv.2014.263.271
INTRODUCTION
Grapevine fanleaf virus (GFLV) is a pathogen of worldwide distribution and the causal agent of the most economically consequential viral disease of grapevines (Auger et al., 1992). The symptoms of GFLV on the leaves can vary from a ‘fanleaf ’ shaped deformation to a yellow mosaic and vein banding (Krake et al., 1999). The virus is transmitted by the Longidorid nematode Xiphinema index Thorne and Allen (Andret-Link et al., 2004). The major means for its long-distance dissemination is by vegetative propagation or grafting of infected materials.
The GFLV is a member of the genus Nepovirus, family Secoviridae (King et al., 2011; Sanfacon et al., 2009). The GFLV genome is composed of two positive single-stranded RNA molecules, encapsidated separately (Mayo and Robinson, 1996). Both genomic RNAs are covalently linked at their 5’end to a small virus-encoded protein (VPg) and are polyadenylated at their 3’end. The 3’Non-Coding Regions (NCR) of many nepoviruses are identical for the two genomic RNAs (Le Gall et al., 1995a, b).
The largest RNA1 (7.3 kb) encodes a polyprotein which is cleaved by an RNA1 encoded viral proteinase into five products, including a proteinase and a polymerase. RNA2 (3.8 kb) has three final in vitro maturation products: The N-terminal 2AHP protein, implicated in the replication of RNA2; the 2BMP putative movement protein and the 2CCP coat protein.
In our previous study with GFLV, the virus was found to be present in cultivated grapevines in the Czech Republic (Kominek, 2008). A mild isolate of the virus, without any obvious fanleaf symptoms, was recorded in South Moravia and was partially characterised at the molecular level (Kominek et al., 2006). Further isolates from South Moravia were characterised at the Horticultural Faculty of Mendel University, Brno (Eichmeier et al., 2010).
In this study, we report on further continuation of our GFLV research the development of a reliable and sensitive method for GFLV detection, suitable for certification of grapevine propagation materials and GFLV field testing. While ELISA is considered to be suitable for routine detection, it is not sufficiently sensitive (Liu et al., 2013). For an evaluation of the health status of grapevines, especially those presented in the screenhouse of Mendel University in Brno, Faculty of Horticulture in Lednice, for use as a nuclear stock for virus-free plants (OEPP/EPPO., 1993), a more sensitive method based on nucleic acid detection would be more appropriate. Primers M2/M3 of Wetzel et al. (2002) were shown to be suitable for our purposes (Eichmeier et al., 2011; Eichmeier, 2014). As these primers can detect both GFLV and ArMV viruses together, the Real-Time RT-PCR system with a newly-designed TaqMan® probe (this study) was used for the specific detection of GFLV. The GFLV detection and quantification was verified in three major phenophases, in field-grown grapevines with different expected levels of viral concentrations. Moreover, the comparison with DAS-ELISA was done upon the same plants and the same samples in order to verify the higher reliability and sensitivity of the Real-Time RT-PCR TaqMan® assay. The end-point dilution of DAS-ELISA was determined, in order to calculate the detection limit of this method for GFLV.
MATERIALS AND METHODS
GFLV-infected plants: Grapevines for the experiment were collected in a field-grown grapevine gene-bank situated within Mendel University in Brno's Faculty of Horticulture in Lednice:
| • | 63TK, cv., Dimrit, originating from Turkey, was provided by Dr. Digiaro, IAM Bari Valenzano, Italy |
| • | 55TK, cv., Cinsaut, originating from Turkey, was provided by Dr. Digiaro, IAM Bari Valenzano, Italy |
| • | UR11, cv., unknown, originating from Russia, was provided by Dr. Digiaro, IAM Bari Valenzano, Italy |
| • | HV5, cv., unknown, was found in a vineyard near Horní Věstonice in the South Moravian region of the Czech Republic and was used in our previous study (Kominek et al., 2006) |
| • | PN32 and PN35, cv., Pamjati Negrula, gene-bank, Faculty of Horticulture, Lednice, Mendel University in Brno |
| • | KML50, cv., Kišmiš Lučistyj, gene-bank, Faculty of Horticulture, Lednice, Mendel University in Brno |
| • | KO1, cv., Kodrjanka, gene-bank, Faculty of Horticulture Lednice, Mendel University in Brno |
Cultivars Kodrjanka, Pamjati Negrula and Kišmiš Lučistyj originated from Moldova as promising genetic materials but they have been grown in the gene-bank in Lednice for more than 20 years.
The presence of the virus in all of the grapevine plants had been confirmed earlier by conventional RT-PCR and sequencing (Eichmeier et al., 2010; Eichmeier, 2014), so we used GFLV verified positive grapevines.
Sample harvest: The isolates were harvested in 2011, in three different phenophases, in three very important phenophases in cultivation season of grapevine in the Czech Republic (climatic conditions of central Europe). The samples for the analyses were obtained precisely on: January 31 (dormancy, BBCH 00), June 6 (from the beginning of flowering to ending of flowering, BBCH 61-68) and October 3 (softening of berries and harvest ripeness, BBCH 83-85); always in the form of cortical scrapings. We used those three phenophases because it is an interesting information about virus concentration in various tissues depending on season and it gives an important recommendation for proper virus detection. The collected samples were thus composed from a mixture of cortex, sclerenchyma, phloem and xylem. The same sample was always used for both the Real-Time RT-PCR TaqMan® and the ELISA detections.
RNA isolation: Total RNA was isolated from the samples and collected using a SpectrumTM Plant Total RNA Kit (Sigma, St. Louis, USA). The isolation was done using 50 mg of plant tissue. The RNA concentration was measured by a fluorometer (Turner BioSystems, Sunnyvale, USA) and the total RNA was adjusted to the level of 50 ng μL-1 for the Real-Time RT-PCR TaqMan® detection.
Reverse transcription: The RT reaction mix consists of 0.25 μg random primer p(dN)6 (Roche, Penzberg, Germany), 0.5 μg of total RNA, plus an equal amount of water (HPLC purity)- for a total volume of 12.5 μL. This mixture was heated in a thermocycler at 95°C for 5 min and then the samples were cooled. Next, 4 μL 5x RT buffer, 1.25 μL of 10 mM dNTPs, 200U M-MLV reverse transcriptase and water were added to make a total volume of 20 μL. The reaction was run on a thermocycler for 60 min at 42°C.
Real-Time PCR TaqMan® assay: The reaction mix consisted of a total volume of 20 μL: The 2 μL of cDNA, 10.85 μL of water (HPLC purity), 4 μL 5x GoTaq®Flexi Buffer (Promega, Fitchburg, USA), 0.5 μL of 10 mM dNTPs (Invitek, Berlin, Germany), 1.25U GoTaq®Hot Start polymerase (5 U μL-1) (Promega, Fitchburg, USA), 1 μL of 10 μM of each of the M2 (YTRGATTTTAGGCTCAATGG) and M3 (TGYAARCCAGGRAAGAAAAT) primers and 0.4 μL of 10 μM TaqMan® probe GF1 (FAM-GATGCGCTAGAAATA-Black Hole Quencher). The temperature program used was: The 94°C for 5 min, followed by 40 cycles of 94°C for 20 sec, 42°C for 20 sec, 72°C for 20 sec and a final extension at 72°C for 7 min. Real-Time analysis was done in a Rotor-GeneTM 3000 thermocycler (Corbett Research, Venlo, Netherlands).
Standard and calibration curve: The PCR product of the GFLV isolate KO1 (size = 293 bp) was inserted into vector pCR4 (Invitrogen, Carlsbad, USA). Subsequently, a standard with a known number of copies (108 copies/μL) was prepared by Generi Biotech Ltd. (Hradec Kralove, Czech Republic). The calibration curve was performed by dilution of the standard within the range of 102-108 copies/μL (diluted by water of HPLC purity).
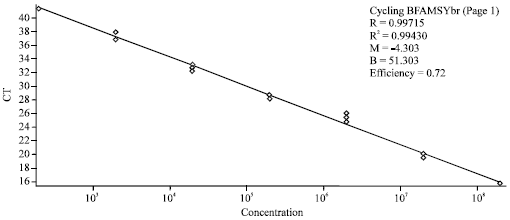 | |
| Fig. 1: | Linear regression curve of plasmid DNA (carrying a GFLV RNA2 insert of KO1 isolate) dilutions in water (HPLC purity), plotted against the logarithm of the estimated concentrations (intra-assay triplicates shown). There are values of threshold cycles CT on the Y-axis and concentration numbers in copies of M2/M3 PCR fragments per microliter on the X-axis |
This calibration scale reflects the range of GFLV positive isolates; with the most robust isolates assigned the value of 107 copies/μL. The lower limit was 102 copies/μL (Fig. 1). All dilutions were tested in triplicate, under the conditions described above. The variability coefficient (%Var) was independently evaluated for all dilutions, within the framestudy of any single run (intra-assay). If the %Var value was 30% or less, it was possible to use it for the quantitative analysis (Burns et al., 2004).
ELISA: Standard DAS-ELISA with GFLV antibodies, purchased from Loewe (Sauerlach, Germany), was used for the virus detection, according to manufacturer's instructions. Samples (0.5 g, identical with those used for RNA isolation) were ground in a sample buffer (composition per 1 L: The 8.0 g NaCl; 2.4 g Tris; 20 g PVP K25; 0.2 g KCl; 0.2 g NaN3; 0.5 mL Tween 20; pH = 7.4), at a ratio of 1:10 (w/V), using a Homex 6 homogenizer (Bioreba, Reinach, Switzerland).
For an estimation of the virus titre, serial dilutions of each tested sample were prepared within the range from 1:10-1:160 000 using the sample buffer. All of the dilution steps were used in the DAS-ELISA in a single microplate. Absorbance data was measured using OpsysMR instrument (Dynex technologies, Bustehrad, Czech Republic). A positive reaction of the ELISA was determined as the mean of the negative samples plus three times the standard deviation (Sutula et al., 1986).
RESULTS AND DISCUSSION
Primers M2/M3, according to Wetzel et al. (2002), were used for the Real-Time RT-PCR detection of GFLV. They are targeted toward a highly conserved part of the 2AHP gene of GFLV (more precisely, 1320-1609 nts). As they can align to ArMV RNA as well, the TaqMan® probe was designed to ensure specific GFLV detection. A highly conserved region was selected because of the fact that different detection results, based on nucleic acid amplification, are primarily influenced by nucleotide polymorphism within the target sequence, or a recombination between closely related viruses such as GFLV and ArMV (Vigne et al., 2008); resulting in the exchange of partial genome sequences which may involve the assay target sequence.
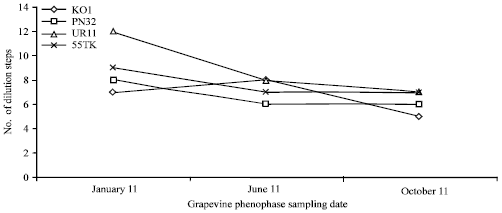 | |
| Fig. 2: | Concentration of GFLV PCR fragments by ELISA and the number of dilution steps, which were positive in ELISA, where a basic dilution (1:10) = 1. Positive reaction in ELISA is equal to three standard deviations above the mean of the negative samples. Highest concentration of viral protein was detected in January 2011 of those isolates |
| Table 1: | Description of measured values in three important phenophases in season |
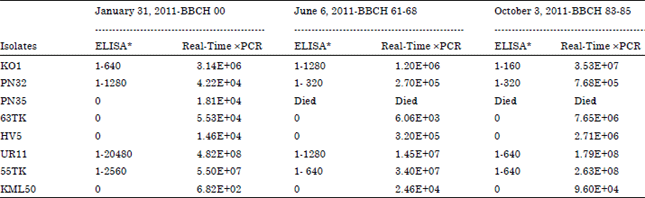 | |
| *End point dilution, x Number of M2/M3 fragment copies per μL, BBCH: Phenophases | |
The efficiency of PCR amplification was estimated to be 72% (on average), the correlation coefficient was R2 = 0.99430 and the characteristics of the curve were y = -4.258x+51.302. Seven tested ranks (102-108 copies/μL) and their appropriate Ct values (15.36-43.94) delimit the range of the GFLV isolates usually measured.
The Real-Time RT-PCR with a TaqMan® probe was used for the estimation of GFLV RNA concentrations in the plant phloem tissues. The test was done on eight GFLV isolates and in three different phenophases. The concentration of GFLV RNA2 molecules was estimated using a calibration curve. The results of the detection and concentration estimations of the eight GFLV isolates in three different phenophases are given in Fig. 2 and 3 and the Table 1. The lowest detected GFLV concentration in our experiments on plant tissues was 6.82x102 molecules of RNA2 in isolate KML50 (Fig. 2). However, the GFLV concentration in the plants never dropped under this level during the tests; all eight isolates were detected in the tissues in all three phenophases; with the exception of isolate PN35 which died shortly after the first sampling. Similarly,Cepin et al. (2010) reported the detection limit of the Real-Time RT-PCR GFLV as 102 copies/μL; they used both relative and absolute quantification with TaqMan® probes.
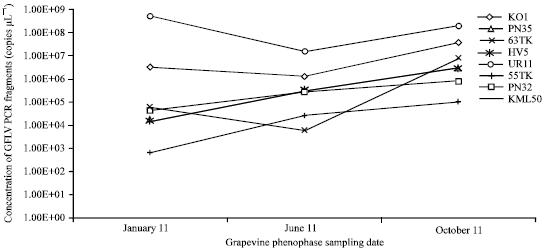 | |
| Fig. 3: | Concentration of GFLV PCR fragments measured by Real-Time RT-PCR TaqMan®. There is clear evidence that Real-Time RT-PCR TaqMan® was able to detect GFLV more precisely than DAS-ELISA |
This is the same level of sensitivity as in our study. In order to compare, the ELISA detection and determination of the end-point dilution was performed from the same samples as were the Real-Time RT-PCR. Based on the Real-Time PCR data, the ELISA-detectable amounts of GFLV particles were estimated. Both methods were compared in their sensitivity as well as in phenophase suitability for GFLV detection. The results of the GFLV detections and concentration assessments, using both methods, are shown in Fig. 2 and 3.
Using the Real-Time RT-PCR method for the determination, the GFLV concentration in the plant tissues had an increasing trend during the year, with the highest values in October. The only exception was for isolate KO1 which had its highest GFLV concentration in January but the October value was its second highest. When looking at the GFLV concentration evaluated by DAS-ELISA, the trend of the virus concentration changes observed by this method were the inverse of those of the Real-Time RT-PCR. ELISA evaluated the highest concentration of GFLV in January. With the ELISA test, only isolate KO1 displayed its highest GFLV concentration in June; however, it was only by one dilution step higher than in January. The lowest GFLV titre in all of the infected plants examined by ELISA was in October.
The M2/M3 primers were not originally designed for the Real-Time PCR method, giving rise to a larger PCR product (290 bp) than is recommended for this method. This was the reason why the amplification efficiency was a little bit lower (72%) than the optimum (100%); however, it is still sufficient (Regier and Frey, 2010).
The GFLV detection using DAS-ELISA showed a lowered sensitivity compared to the Real-Time RT-PCR; with 4 out of the 8 tested GFLV isolates not being detected in any of the three phenophases.
These four ELISA-negative isolates indicated concentrations of 5.53x104 viral particles (and lower), based on the parallel Real-Time RT-PCR data. Cepin et al. (2010) stated that ELISA (Bioreba, Nylon, Switzerland) is able to detect a concentration of 105 copies/μL and this was confirmed by our results, although in our experiment we used another supplier of ELISA reagents (Loewe).
The contradictions observed between the results of the two methods may be explained by the fact that the GFLV coat protein gene is expressed less in autumn phenophases, or by an overproduction of viral RNA at that time (Cepin et al., 2010). However, this hypothesis should be further studied in future research.
Cepin et al. (2010) and Rowhani et al. (1992) reported that the summer period is the worst for GFLV detection. Our results showed a relatively stable concentration of GFLV in grapevine phloem tissues over the course of a year. Summer time (June 2011) was generally a little weaker in the GFLV titre but it was still detectable by both methods (Real-Time RT-PCR and DAS-ELISA). For this reason we cannot conclude as to which sampling term is the best for routine testing of GFLV presence when using phloem, every time in the course of a year should be suitable for GFLV detection by both methods tested. However, the differences in the sensitivities of the method used for virus detection (DAS-ELISA or Real-Time RT-PCR) as was demonstrated in our research must be taken into consideration.
Interesting results were found in the case of isolate HV5. When it was collected in the field (Kominek et al., 2006), it was easily detectable by ELISA. After several years of cultivation in a gene-bank (screenhouse), the concentration of GFLV decreased to under the detection limit for ELISA. An understanding of this phenomenon is also needed in future research.
CONCLUSION
Based on our results we established the best timing for sampling to detecting GFLV occurrence by Real-Time RT-PCR TaqMan® and DAS-ELISA.
The first method Real-Time RT-PCR was effective for detection of all isolates which were included in the examined set of potentially GFLV positive plants. Utilizing this method, the number of copies per microliter was determined as most abundant in the last phenological phase, BBCH 83-85-softening of berries and harvest ripeness (October 3, 2011 in the Czech Republic). Between the phenophases BBCH 00 a BBCH 61-68 clear correlation of increase or decrease of concentration was not confirmed. In conclusion, it can be determined that the best phenological stage for the detection of GFLV by RT-PCR with primers M2/M3 and TaqMan probe GF1 is phenophase BBCH 83-85.
By means of the second determination method it was possible to detect only 4 positive isolates with the highest copy concentration per microliter (KO1, 55TK, PN32 and UR11), described in Table 1. Unlike the first method, the first phenophase (BBCH 00) was found as the most appropriate for GFLV detection and vice versa BBCH 83-85 was the least suitable. Using ELISA method, the presence of GFLV was impossible to determine in PN35, 63TK, HV5 and KML50 isolates. Finally, it can be established that the sera used in the ELISA testing are limiting in relation to Real Time RT-PCR detection of GFLV presence for concentration as low as 10E+05 copies per microliter of PCR product approximately.
ACKNOWLEDGMENTS
This study was supported by the Grants No. MZe 0002700604 and No. QH91214 of the Ministry of Agriculture of the Czech Republic. The authors wish to thank Peter Lemkin and Zuzana Mynarzova for editing of the manuscript.
REFERENCES
- Andret-Link, P., C. Schmitt-Keichinger, G. Demangeat, V. Komar and M. Fuchs, 2004. The specific transmission of Grapevine fanleaf virus by its nematode vector Xiphinema index is solely determined by the viral coat protein. Virology, 320: 12-22.
CrossRefDirect Link - Auger, J., E.E. Aballay, C.M. Pinto and V.C. Pastenes, 1992. Effect of Grape Fanleaf Virus (GFV) on growth and productivity of grapevine plants cv. Thompson Seedless. Fitopatologia, 27: 85-89.
Direct Link - Burns, M.J., H. Valdivia and N. Harris, 2004. Analysis and interpretation of data from real-time PCR trace detection methods using quantitation of GM soya as a model system. Anal. Bioanalyt. Chem., 378: 1616-1623.
CrossRefPubMedDirect Link - Cepin, U., I. Gutierrez-Aguirre, L. Balazic, M. Pompe-Novak, K. Gruden and M. Ravnikar, 2010. A one-step reverse transcription real-time PCR assay for the detection and quantitation of Grapevine fanleaf virus. J. Virol. Methods, 170: 47-56.
CrossRefPubMedDirect Link - Eichmeier, A., M. Baranek and M. Pidra, 2010. Analysis of genetic diversity and phylogeny of partial coat protein domain in Czech and Italian GFLV isolates. Plant Prot. Sci., 46: 145-148.
Direct Link - Eichmeier, A., 2014. Sequence differences between Grapevine fanleaf virus and Arabis mosaic virus isolates in RNA2 encoding the central part of movement protein. Int. J. Virol., 10: 243-252.
Direct Link - Kominek, P., M. Bryxiova and M. Glasa, 2006. Partial molecular characterization of a mild isolate of Grapevine fanleaf virus from South Moravia, Czech Republic. Acta Virol., 50: 201-205.
PubMed - Le Gall, O., T. Candresse and J. Dunez, 1995. Transfer of the 3' non-translated region of grapevine chrome mosaic virus RNA-1 by recombination to tomato black ring virus RNA-2 in pseudorecombinant isolates. J. Gen. Virol., 76: 1285-1289.
CrossRefDirect Link - Le Gall, O., T. Iwanami, A.T. Jones, K. Lehto and H. Sanfacon et al., 1995. Comoviridae. In: Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses. Fauquet, C.M., M.A. Mayo, J., Maniloff, U. Desselberger and L.A. Ball (Eds.). Elsevier Academic Press, London, UK., pp: 807-818.
- Liu, W., X. Zhao, P. Zhang, T.T. Mar and Y. Liu et al., 2013. A one step real-time RT-PCR assay for the quantitation of Wheat yellow mosaic virus (WYMV). Virol. J., Vol. 10.
CrossRef - Regier, N. and B. Frey, 2010. Experimental comparison of relative RT-qPCR quantification approaches for gene expression studies in poplar. BMC Mol. Biol., Vol. 11.
CrossRefDirect Link - Rowhani, A., M.A. Wlaker and S.S. Rokni, 1992. Sampling strategies for the detection of grapevine fanleaf virus and the grapevine strain of tomato ringspot virus. Vitis, 31: 35-44.
Direct Link - Sanfacon, H., J. Wellink, O. Le Gall, A. Karasev, R. van der Vlugt and T. Wetzel, 2009. Secoviridae: A proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus and the proposed genus Torradovirus. Arch. Virol., 154: 899-907.
CrossRefDirect Link - Sutula, C.L., J.M. Gillett, S.M. Morrissey and D.C. Ramsdell, 1986. Interpreting ELISA data and establishing the positive-negative threshold. Plant Dis., 70: 722-726.
CrossRef - Vigne, E., A. Marmonier and M. Fuchs, 2008. Multiple interspecies recombination events within RNA2 of Grapevine fanleaf virus and Arabis mosaic virus. Arch. Virol., 153: 1771-1776.
CrossRefDirect Link - Wetzel, T., R. Jardak, L. Meunier, A. Ghorbel, G.M. Reustle and G. Krczal, 2002. Simultaneous RT/PCR detection and differentiation of arabis mosaic and Grapevine fanleaf nepoviruses in grapevines with a single pair of primers. J. Virol. Methods, 101: 63-69.
CrossRefDirect Link