
Paul C. Chikezie
Department of Biochemistry, Imo State University, Owerri, Nigeria
Okey A. Ojiako
Department of Biochemistry, Federal University of Technology, Owerri, Nigeria
Agomuo C. Ogbuji
Department of Food Science and Technology, Abia State Polytechnic, Aba, Nigeria
International Journal of Biological Chemistry
Year: 2015 | Volume: 9 | Issue: 3 | Page No.: 92-109







ABSTRACT
Oxidative stress is the outcome of an imbalance between the production and neutralization of reactive oxygen and nitrogen species (RONS) such that the antioxidant capacity of cell is overwhelmed. The present review briefly summarized the underlying role of overwhelming levels of RONS in the pathophysiology of diabetes mellitus (DM). The review is based on using keywords to obtain information from publications in PubMed, ScienceDirect and Google Scholar from 1970-2015. The primary causative factor of oxidative stress in DM is hyperglycemia, which operates via several mechanisms. However, the individual contribution of other intermediary factors to hyperoxidative stress remains undefined, in terms of the dose response relationship between hyperglycemia and overall oxidative stress in DM. Intuitively, the inhibition and/or scavenging of intracellular free radical formation provide a therapeutic strategy to prevent oxidative stress and ensuing pathologic conditions. The integration of antioxidants formulations into conventional therapeutic interventions, either by ingestion of natural antioxidants or through dietary supplementation should be encouraged for a holistic approach to the management and prevention of DM and complications associated with the pathology.
PDF Abstract XML References Citation
Received: May 21, 2015;
Accepted: June 30, 2015;
Published: August 06, 2015
How to cite this article
Paul C. Chikezie, Okey A. Ojiako and Agomuo C. Ogbuji, 2015. Oxidative Stress in Diabetes Mellitus. International Journal of Biological Chemistry, 9: 92-109.
URL: https://scialert.net/abstract/?doi=ijbc.2015.92.109
URL: https://scialert.net/abstract/?doi=ijbc.2015.92.109
INTRODUCTION
Oxidative stress is the outcome of an imbalance between the production and neutralization of reactive oxygen and nitrogen species (RONS) such that the antioxidant capacity of cell is overwhelmed (Shin et al., 2001; Styskal et al., 2012; Sellamuthu et al., 2013; Poljsak and Fink, 2014). Ordinarily, the peculiar molecular configuration of oxygen (O2) confers a very slow reactivity between O2 and biomolecules. Two main factors make O2 kinetically insert; the spin restriction imposed by its triplet state and the negative standard potential for one electron reduction of O2 to superoxide radical (O2•–). However, O2 possesses the attributes of free radicals in that it has two unpaired electrons with parallel spin in different -anti-bonding orbitals that is responsible for its paramagnetic properties and relative stability (Pollack and Leeuwenburgh, 2000; Poljsak and Fink, 2014). Spin restriction can be overcome by single electron exchange that converts O2 to strong oxidizing agent (Rotilio et al., 2000; Thannickal and Fanburg, 2000). Therefore, the activation of O2 by specific enzymes is achieved by the presence, at the active site, of either flavins or reduced transition metals such as iron (Fe2+) and copper (Cu2+), which donates single electron to O2 (Rotilio et al., 2000). In the case of metalloproteins, a varying degree of electron transfer from the metallic moiety to O2 is possible. On this basis, metalloproteins can behave either as O2 carriers (hemoglobin, hemocyanin, hemerythrin, myoglobin), where reversible interaction with O2 occurs, or as O2 reductants. Studies showed that autoxidation of oxy-hemoglobin elicit the generation of free radicals (Moussa, 2008).
Electron transfer to O2 is catalyzed by oxidases for production of chemical energy or oxidation of substrates. These enzymes, located in different subcellular compartments (mitochondria, endoplasmic reticulum, peroxisomes) are potential sources of partially reduced Cu2+ derivatives in biological milieu. Cytosolic enzymes {xanthine oxidase, NADPH oxidases, lipoxygenase, cyclooxygenase (COX), cytochrome P450 enzymes and aldehyde oxidase}, uncoupled endothelial nitric oxide synthase (eNOS) and other hemoproteins also produce O2•– during catalysis (Yung et al., 2006; Alfadda and Sallam, 2012; Styskal et al., 2012). The mitochondrial electron transport chain reduces O2-O2•– at ubiquinone and NADH dehydrogenase sites, whereas; microsomal cytochrome P450 and its reductases produce O2•– during xenobiotic biotransformation (Desco et al., 2002; Wright et al., 2006; Sugatani et al., 2006; Bajaj and Khan, 2012). The "Leaky" inner mitochondrial membrane electron transport chain directly reacts with O2 to generate O2•–, which dismutase to form hydrogen peroxide (H2O2), which can further react to form the hydroxyl radical (•–OH) (Pollack and Leeuwenburgh, 2000; Alfadda and Sallam, 2012; Styskal et al., 2012). Additionally, the mitochondrial outer membrane enzyme, monoamine oxidase, catalyzes the oxidative deamination of biogenic amines and it’s a quantitatively large source of H2O2 that contributes to increase in steady state concentrations of reactive species within both the mitochondrial matrix and cytosol (Cadenas and Davies, 2000). Specifically, O2•– is the primary radical formed by the reduction of O2 leading to secondary radicals or reactive oxygen species (ROS) such as H2O2 and •–OH in the mitochondria (Pollack and Leeuwenburgh, 2000; Styskal et al., 2012). Although, the cause-effect relationship remains tentative, there appears to be a strong association between mitochondrial dysfunction and chronic metabolic diseases such as Type II diabetes mellitus (T2DM) and obesity (Alfadda and Sallam, 2012). The origin, enzymatic pathways of ROS and their oxidized products, as well as their enzymatic inactivation pathways in T2DM have previously been summarized (Hayden and Tyagi, 2004).
The RONS have been implicated in the pathophysiology of various disease states, including diabetes mellitus (DM) and long-term development of associated complications (Hayden and Tyagi, 2004; Wright et al., 2006; Giacco et al., 2010; Alfadda and Sallam, 2012; Bajaj and Khan, 2012). Oxidative tissue damage is mediated by activating a number of cellular stress-sensitive pathways, which include nuclear factor-ĸB (NF- ĸB), p38 mitogen-activated protein kinase, NH2-terminal Jun kinases/stress-activated protein kinases and hexosamines (Evans et al., 2003). Consequently, imbalance between cellular generation and scavenging capacity of free radicals elicits tissue damage associated with DM pathology (Betteridge, 2000; Bajaj and Khan, 2012; Styskal et al., 2012). Also, incidents of oxidative stress-induced neurological disorders mediated by inhibition of enzymatic activities connected with neurotransmission have been reported in experimental diabetic rats (Ashokkumar et al., 2006; Ghareeb and Hussen, 2008; Alipour et al., 2012). As a follow up to these findings, it is obvious that understanding the relationship between oxidative stress and DM pathology has the potentials to expand the therapeutic intervention options against the pathogenesis and progression of the disease. Therefore, the present review briefly summarized the underlying role of overwhelming levels of RONS in the pathophysiology of DM. The review is based on using keywords to obtained information from publications in PubMed, ScienceDirect and Google Scholar from 1970-2015.
Oxidative damage and modification of macromolecules: The radicals (O2•–, •–OH, NO‾, 1O2, RO‾2, ‾ONOO) and pro-radicals (H2O2, HOCl, RS and O3) are extremely reactive molecules. In biological systems, RONS cause substantial damage/modification to functional and structural macromolecules (lipids, nucleic acids and proteins), as well as modulation of activity of antioxidant enzymes (Poljsak and Fink, 2014). Oxidative attack of polyunsaturated fatty acids (PUFAs) gives rise to peroxided molecules, which subsequently breakdown to form reactive metabolites. For the fact that lipids are the major components of biological membranes, fluidity and permeability of these supra-molecules are severely affected, together with membrane protein functionality (Poljsak and Fink, 2014). The reactive aldehydes are cytotoxic products of lipid peroxidation. Specifically, 4-hydroxynonenal (HNE) causes long-lasting biological consequences by covalent modification of macromolecules, whereas at physiological levels, HNE is considered as second messengers of free radicals and signaling molecules. Report showed that HNE and related reactive aldehydes may play critical roles in the pathophysiology of DM, in terms of the pathogenesis, progression and complications of the disease (Jaganjac et al., 2013).
Base modification, scission of deoxyribose rings, strand breaks and ultimately, chromosomal aberration are outcomes of oxidative damage to nucleic acids. Oxidative challenge on proteins leads to the modification of amino acids side chains with the introduction of carbonyl groups, or oxidation of sulphydryl groups with consequent cross linking and aggregation of protein molecules. The presence of oxidative modifications ultimately results in increased susceptibility of modified proteins to specific proteases, enzyme deactivation, or conversely, unwarranted activation of enzymes (Desco et al., 2002; Poljsak and Fink, 2014).
There appears to be a direct mechanistic link between oxidative stress and the etiology of DM through the accumulation of oxidative damage to critical macromolecules. Several studies have established an association between increased carbonylation and nitrosylation of proteins in insulin-sensitive tissues and T2DM (Kaneki et al., 2007; Grimsrud et al., 2008; Muellenbach et al., 2008). In another study, evidence showed that oxidation of specific proteins compromised their function in vitro (Levine, 1983; Levine et al., 1999) and there is a correlation between increasing oxidative stress and diminished protein folding and function in different animal models (Pierce et al., 2008; Perez-Matute et al., 2009).
Oxidative stress is as a result of free radicals generated during autoxidation of glucose in DM (Aronson and Rayfield, 2002; Evans et al., 2003). Overall, DM is characterized by raised level of oxidative stress with associated increased generation of glycoxidation products, notably, HbA1c above the benchmark plasma value <7% (Hayden and Tyagi, 2004; Wright et al., 2006; El-Wassef et al., 2012). The presence of hyperglycemia promotes increase in intracellular levels of advanced glycation end products (AGEs) (Wolf and Ziyadeh, 2007; Di Naso et al., 2011; Musabayane, 2012). Furthermore, auto-oxidation of glucose generates ROS, such as O2•–, H2O2 and •–OH (Bajaj and Khan, 2012; Moussa, 2008), which in turn, accelerate lipid peroxidation with corresponding accumulation of advanced lipoxidation end products (ALEs) and more free radicals (Rolo and Palmeira, 2006; Jaganjac et al., 2013). Increased levels of ROS in T2DM also contribute to a hypercoagulable state and evidence suggests that accumulation of oxidation products occur prior to the development of DM (Matteucci and Giampietro, 2000).
Antioxidants such as the flavonoids prevent the formation of AGEs by impeding the glucose dependent formation of Amadori, Schiff bases or Milliard products, which are intermediary products leading to the formation of AGEs (Keaney and Loscalzo, 1999; Musabayane, 2012). Likewise, disruptions of AGEs cross linkages by drugs such as alagebrium or inhibition of AGE signal transduction pathway can substantially prevent the accumulation and formation of AGEs, respectively (Hartog et al., 2007). The option of shielding or obliteration of AGEs’ receptor (RAGE), expression of RAGE antisense cDNA or anti-RAGE ribozyme may reverse atherosclerosis in experimental animals (Ihara et al., 2007; Giacco et al., 2010). Also, notable inhibitors (amino guanidine and pyridoxamine) of AGEs formation exhibit reno-protective effects in diabetic animals (Lassila et al., 2004; Hartog et al., 2007).
Mechanisms of hyperglycemia induced production of oxygen free radicals: Hyperglycemia is known to cause elevation in plasma free radical concentrations (Hammes et al., 1997; Cimato et al., 2008). The production of free radicals is engendered by uncontrolled hyperglycemia, which may occur via several routes (Rolo and Palmeira, 2006; Giacco et al., 2010; Bajaj and Khan, 2012): (1) increased glycolysis (Vaag et al., 1992), (2) intercellular activation of sorbitol (polyol) pathway (Williamson et al., 1993; Di Naso et al., 2011), (3) autoxidation of glucose (Wolff et al., 1991), (4) protein kinase C (PKC) dependent activation of NAD(P)H oxidase (Inoguchi et al., 2003), (5) increased hexosamine pathway flux (Rolo and Palmeira, 2006), (6) increased intracellular formation of AGEs (Giacco et al., 2010), (7) increased expression of receptor for AGEs and its activating ligands (Giacco et al., 2010) and (8) non-enzymatic protein glycation (Ceriello et al., 1992). The overall rate of formation of oxidative products leading to oxidative tissue damage is summarized in Fig. 1.
Hyperglycemia appears to enhance non-oxidative catabolism of glucose to lactate, which is associated with increase in NADH/NAD+ ratio (Vaag et al., 1992; Williamson et al., 1993). Under the condition of accelerated glycolysis, oxidation of glyceraldehyde 3-phosphate (GAP) to 1, 3-biphosphoglycerate (1, 3-DPG) by glyceraldehyde 3-phosphate dehydrogenase is coupled to reduction of NAD+ to NADH and appears to become the rate limiting step in glycolysis (Kobayashi and Neely, 1979). In the cytosol, NADH is oxidized to NAD+ by lactate dehydrogenase (LDH) with concomitant reduction of pyruvate to lactate.
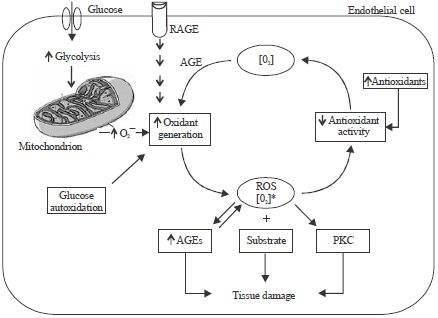 | |
| Fig. 1: | Relationship between rates of oxidant generation, antioxidant activity, oxidative stress and oxidative damage in diabetes, RAGE: Receptor for AGEs (Aronson and Rayfield, 2002) |
Thus, increase in the ratio of NADH/NAD+ reflects increased lactate/pyruvate ratio (Williamson et al., 1993). The mechanism by which increased rate of glycolysis increases free cytosolic NADH/NAD+ ratio (redox imbalance) suggest disequilibrium between the rate of oxidation of GAP to 1, 3-DPG and the rate of reduction of pyruvate to lactate (Kobayashi and Neely, 1979). Thus, enhanced glycolysis as a result of hyperglycemia is associated with increase in NADH/NAD+ ratio due to impaired oxidation of NADH to NAD+.
The increase in glucose flux via sorbitol pathway (a pathway of a minor significant under normal glycemic condition) elicits one of the major metabolic disturbances associated with diabetic hyperglycemia (Ciuchi et al., 1996). In this pathway, glucose is reduced to sorbitol by aldose reductase (AR) coupled with oxidation of NADH/NAD+ (Dallak et al., 2008). Subsequently, sorbitol is oxidized to fructose by NADH dependent sorbitol dehydrogenase (SDH) (Cameron et al., 1997; Giacco et al., 2010). Previous studies have suggested several hypotheses for tissue injury engendered by increased sorbitol pathway activity, thus:
The decreased availability of NADPH, which is required for maintenance of reduced glutathione (GSH), is oxidized to NADP+ by the reduction of glucose to sorbitol by AR pathway (Tilton et al., 1995). Furthermore, the competition between AR and glutathione reductase (GSH-R) for NADPH cofactor further depletes intracellular GSH (Ciuchi et al., 1996). Attention has been focused on GSH depletion, because it dictates levels of cellular ROS production and accumulation, which in turn have a bearing on extent of oxidative tissue damage in DM (Brownlee, 1994). Increased ratio of NADH/NAD+ is connected with accelerated oxidation of sorbitol to fructose by NADH dependent SDH (Tesfamariam and Cohen, 1992; Brownlee, 2001). Consequently, NADH molecules generated in the cytosol by oxidation of sorbitol to fructose are eventually conveyed to the mitochondria and oxidized by respiratory chain reaction that result in production of O2•– and other ROS (Williamson et al., 1993; Ceriello et al., 1996). Thus, an increase in the cytosolic NADH may be accompanied by increased load of mitochondrial NADH, which in turn, leads to increased ROS generation.
In a cell-free system under physiological conditions, glucose can be auto-oxidized to H2O2, through enediol tautomer formation, which elicits the accumulation of reactive intermediate such as •–OH and O2•– and ketoaldehydes (Brownlee et al., 1988; Packer, 1993). Transition metals such as Fe2+ promote auto-oxidation of glucose and therefore, are crucial in these reaction cascades (Packer, 1993). Several studies have equally shown that auto-oxidation of glucose in this manner are responsible for increased levels of ROS in DM (Monnier, 1990; Santini et al., 1997).
Non-enzymatic glycation is a spontaneous reaction between glucose and amino groups of proteins in which reversible Shift bases and more stable Amadori products are formed (Aronson and Rayfield, 2002). The AGEs are produced by auto-oxidation of Amadori product (Keaney and Loscalzo, 1999; Ahmed, 2005; Rolo and Palmeira, 2006). Glucotoxicity is elicited through the binding of AGEs to RAGEs, which have been identified in endothelial cells, monocots/macrophages, mesangial cells, neurons and smooth muscle cells (Aronson and Rayfield, 2002; Inoguchi et al., 2003; Hayden and Tyagi, 2004; Yonekura et al., 2005; Wright et al., 2006). The presence of AGEs elicits poor matrix protein flexibility as a result of formation of cross-links among extracellular matrix proteins, which leads to abnormal interactions with other matrix components (Yonekura et al., 2005). Additionally, the interaction of AGEs with endothelial surface RAGEs promote intracellular oxidative stress via the activation of AR of polyol-sorbitol pathways, activation of PKC isoforms and transforming growth factor-β (TGF-β) as well as activation of nuclear factor (NF-ĸB) (Aronson and Rayfield, 2002; Evans et al., 2003). The activation of NF-ĸB promotes increase in expression of a variety of cytokines such as tumor necrosis factors (TNF-α and TNF-β), interleukins (IL) 1, 6, 8 and 18 and interferon-γ, even in the presence of intact antioxidant mechanisms, which may engender overt diabetic nephropathy with associated glomerulosclerosis (Esposito et al., 2002; Aronson and Rayfield, 2002; Inoguchi et al., 2003; Hayden and Tyagi, 2004; Basta et al., 2004; Wright et al., 2006; Styskal et al., 2012).
Also, increased cellular uptake of glucose stimulates PKC activity (Lee et al., 1989) which, amongst other effects, activates peroxidase enzymes and the COX pathway (Lee et al., 1989; Feener and King, 1997; Golbidi et al., 2012), with resultant overproduction of RONS. The process leading to this pathology is further enhanced and amplified, when antioxidant defense mechanisms are compromised (Bierhaus et al., 1997).
Mechanisms of hyperinsulinemia induced production of oxygen free radicals: Decline in physical fitness, increase in body fatness and upper body fat distribution are frequently associated with hyperinsulinemia and insulin resistance (DeFronzo and Ferrannini, 1991). Several lines of evidence indicated that hyperinsulinemia promoted the generation of free radicals by NADPH-dependent mechanism, which involved the activation of phosphatidylinositol 3'-kinase and stimulation of proliferative extracellular signal-regulated kinases (ERK-1- and ERK-2)-dependent pathways (Ceolotto et al., 2004). Furthermore, Krieger-Brauer and Kather (1992) reported that prolong exposure of human adipocytes to insulin caused a time- and dose-dependent accumulation of H2O2 in vitro. This effect, which has been linked to the presence of a membrane-bound NADPH oxidase, was observed to persist after cell disruption and devoid of ATP utilization; an indication that the receptor-kinase activity step was bypassed. In addition, increased insulin concentration in rats following intra-peritoneal injection of dextrose has been reported to be associated with increased free radical production (Habib et al., 1994).
Fasting hyperinsulinemia is considered to be a hallmark of insulin resistance (DeFronzo and Ferrannini, 1991) and there is a relationship between insulin resistance and plasma free radical concentration (Ceriello, 1995, 2000). Factors that contribute to the elevation of free radicals and pathogenesis of insulin resistant DM are as follows:
| • | Hyperinsulinemia overdrive of the sympathetic nervous system (Rowe et al., 1981). Specifically, catecholamine increases free radical production through induction of metabolic rate and auto-oxidation pathway in DM (Singal et al., 1983) |
| • | Insulin resistance is associated with elevated fasting plasma non-esterified fatty acid (NEFA) concentration (DeFronzo and Ferrannini, 1991; Randle et al., 1994) |
Toborek and Henning (1994) showed that NEFA caused raised levels of oxidative stress in cultured endothelial cells following initial decreased level of GSH after 6h of incubation. It is worthwhile to note that the complexity of these multitudes of findings suggests that the generation of free radicals may represent a potential mechanism by which chronic hyperinsulinemia activates proliferative events and down-regulates metabolic signals (Ceolotto et al., 2004).
Oxidative stress induced lipid peroxidation in diabetes mellitus: Lipid peroxidation has been implicated in the pathogenesis of many degenerative disorders (Armstrong et al., 1982) including naturally occurring and chemically induced DM (Rerup, 1970; Nishigaki et al., 1981; Higuchi, 1982). Lipid peroxidation is the primary cellular damage resulting from free radical reactivity of which cellular lipid structures are mostly affected (Toborek et al., 1992; Ahmed, 2005). Oxidative deterioration of PUFAs of cellular membrane phospholipids, via intermediate radical reactions involves the production of hydroperoxides (Rungby et al., 1992; Cameron et al., 1994). The chain reactions are associated with the generation of highly toxic peroxyl radicals (RO2‾) in a cycle of reactions that generate new lipid hydroperoxides (LHP) because of the proximity of PUFAs in biomembranes (Kajanachumpol et al., 1997; Betteridge, 2000).
Also, both radical and non-radical oxidants can induce lipid peroxidation in lipoproteins, particularly those that contain PUFAs. For instance, peroxynitrite (‾ONOO) is particularly a powerful oxidant of low-density lipoproteins (LDL) (Violi et al., 1999). Similarly, in vitro studies have revealed the presence of oxidized LDL (ox-LDL) fractions with identifiable auto-antibodies against ox-LDL in plasma of Type I DM (T1DM) patients, which suggest that the oxidation LDL can as well occurs in DM in vivo (Jain et al., 1998). Accordingly, Maejima et al. (2001) noted raised levels of ‾ONOO in T2DM patients. Additionally, LDL receptor does not recognize ox-LDL and are subsequently taken up by scavenger receptors in macrophages to form foam cells, which leads to atherosclerotic plaques (Boullier et al., 2001; Aronson and Rayfield, 2002).
Early evidence that suggested lipid peroxidation in DM was reported by Sato et al. (1979), in which they noted that the levels of lipid peroxides in plasma of DM patients were significantly higher than that of normal subjects. Likewise, levels of plasma lipid peroxides of DM patients with angiopathy were relatively higher than that of DM patients. They further inferred that raised level of lipid peroxides was among other several factors that initiates atherosclerosis in DM. In another study, Davison et al. (2002) used electron spin resonance (ESR) spectroscopy in conjunction with alpha-phenyl-tert-butylnitrone spin trapping to measure pre- and post-exercise free radical concentration in the venous blood of young male patients suffering from T1DM in order to ascertain their susceptibility to rest and exercise-induced oxidative stress. They suggested that greater concentration of oxidants and LHP were as a result of glucose auto-oxidation couple with lower rate of exercise-induced oxidation of major lipid soluble antioxidant; α-tocopherol in DM. Furthermore, they noted that ESR-detected radicals, in the course of the investigation, were secondary species derived from decomposition of LHP, which were major initial reaction products following free radical attack on biomembranes.
The underlying mechanisms of the formation of LHP and biologically active metabolites, together with their effect on cellular structure and function are becoming of increasing importance in understanding the pathogenesis and management of DM (Crabbe, 1987). For instance, lipoxygenase products, especially 12(S)-HETE and 15(S)-HETE, are involved in the pathogenesis of several diseases including DM (Bajaj and Khan, 2012). The LHPs are produced from a variety of PUFAs precursors via intermediate radical reactions involving O2 and metal cations (Fe2+ and Cu2+). The reactions generate highly reactive and cytotoxic lipid radicals. Extracellular LHP are transported in the systemic circulation by low- and high-density lipoproteins (Nishigaki et al., 1981). When released locally, LHP elicits structural damage to variety of biomolecules. For instance, the formation of LHP and their metabolites are important in ophthalmic medicine in that the retinal portion of eye is particularly sensitive to oxidative stress. Additionally, a steady irreversible decline in electroretinogram is observed in streptozotocin (STZ)-induced diabetic rats (Pautler and Ennis, 1980), when synthetic LHP was injected into the vitreous chamber of experimental animals (Armstrong et al., 1982). Fortunately, LHP induced oxidative damage to biomolecules is ameliorated by lipid and water-soluble antioxidants, as well as by specific antioxidant enzymes.
Oxidative stress indicators in diabetes mellitus: The concept of raised level of oxidative stress (increased generation of free radicals) in DM was derived principally from in vitro experiments (Wolff, 1993; Schiekofer et al., 2003; Wright et al., 2006). One of such investigations involved the use of cultured human umbilical vein endothelial cells incubated in variable glucose concentrations followed by monitoring the generation of ROS by a measure of cellular level of nitrotyrosine (Quagliaro et al., 2003; Wright et al., 2006).
Early observations have focused attention in understanding underlying mechanisms that may be relevant to atherogenesis in patients suffering from T2DM and in obesity. Persons suffering from T2DM and/or obese individual exhibit raised level of oxidative stress and inflammatory response (Jorns et al., 1999; Alfadda and Sallam, 2012), which from reports have been linked to increased cellular levels of inflammatory cytokines, TGF-β and insulin-like growth factor binding protein (IGFBP)-3 (Jorns et al., 1999; Wright et al., 2006; Bajaj and Khan, 2012). Raised level of oxidative stress in T2DM is indicated by an increase in ROS generation by circulating mononuclear cells, increased lipid peroxidation (Nishigaki et al., 1981), protein carbonylation (Aljada et al., 1995), nitro-tyrosine formation (Aydin et al., 2001) and DNA damage (Dandona et al., 1996; Shin et al., 2001; El-Wassef et al., 2012; Styskal et al., 2012). Importantly, even pre-DM individuals showed elevated 8-hydroxyguanosine, which suggested that oxidative damage to DNA is present even before the clinical development of DM (Styskal et al., 2012). Recently, raised level of oxidative stress was also demonstrated in the obese as reflected in increased lipid peroxidation, protein carbonylation and ortho-tyrosine and meta-tyrosine formation in DM individuals (Keaney and Loscalzo, 1999; Cumaoglu et al., 2007; Cimato et al., 2008; Chis et al., 2009; Styskal et al., 2012). However, the levels of these oxidative stress indicators, as well as generation of ROS by leucocytes, were reversed following restriction to 1,000 calories/day for 4 weeks (Dandona et al., 2001).
The primary causative factor of oxidative stress in DM is hyperglycemia, which operates via, several mechanisms (Fig. 2). However, the individual contribution of other intermediary factors to hyperoxidative stress remains undefined, in terms of the dose response relationship between hyperglycemia and overall oxidative stress in DM.
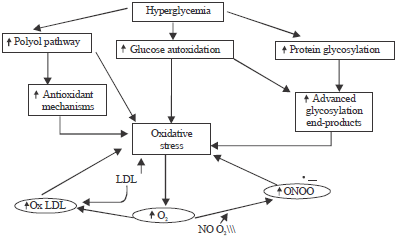 | |
| Fig. 2: | Pathogenesis of hyperoxidative stress in non-insulin dependent diabetes. In boxes are shown mechanisms that are directly related to hyperglycemia. In circles are some mechanisms that result from the reaction of free radicals e.g. superoxide (O2•–) with lipoproteins (e.g. small, dense low- density lipoprotein) and nitric oxide (NO‾), oxidized LDL (ox-LDL), peroxynitrite (‾ONOO) |
In the presence of elevated calcium levels in endothelial cell, hyperglycemia stimulates the synthesis of NO‾ (Cohen, 1993; Poston and Taylor, 1995), in which in the presence of O2•–, NO‾ is converted to highly potent oxidant ‾ONOO that promotes endothelial cell damage and endothelial dysfunction (Beckman et al., 1990; Landmesser et al., 2003). Hyperglycemia causes paradoxical increase in the generation of NO‾ but low availability of NO‾ (Santilli et al., 2004; Wright et al., 2006), which appears to activate NF- ĸB and thereby engendering increased expression of inducible nitric oxide synthase (iNOS) (Spitaler and Graier, 2002). However, Santilli et al. (2004) noted that low availability of NO‾ is attributable to uncoupling of receptor-mediated signal transduction (El-Missiry et al., 2004) and is the primary causative factor of endothelial dysfunction and diabetic angiopathy. In addition, overwhelming levels of O2•– directly, inactivates two critical anti-atherosclerotic enzymes (eNOS and prostacyclin synthase) and consequently, precipitate defective angiogenesis (Giacco et al., 2010).
Although, there are extreme difficulties in measuring free radicals in vivo, some evidence in support of the notion of raised level of oxidative stress in DM and its association with poor metabolic control and coronary heart disease has been derived from observations in patients with DM (Griffin et al., 1997). Raised level of oxidative stress may provide a plausible pathophysiologic basis for the direct link between hyperglycemia and increased cardiovascular risk in DM (Lehto et al., 1997). There is persuasive evidence and definitive clinical proof that oxidative stress is associated with the pathogenesis and progression of atherosclerosis in both diabetic and non-diabetic subjects (Aronson and Rayfield, 2002). Insulin resistance and raised level of oxidative stress have been observed in obese T2DM patients (Skrha et al., 1996).
There is a relationship between plasma malondialdehyde (MDA) concentration and hyperglycemia (Hayden and Tyagi, 2004; Chikezie and Uwakwe, 2014). Earlier reports by Sato et al. (1979) noted increased level of TBARS in blood samples of patients with poorly controlled DM and diabetic angiopathy. The elevation in TBARS concentration is considered to be an indicator of marked organ or tissue degeneration (El-Missiry et al., 2004). Also, elevation of TBARS concentration provides an indirect measurement of level of lipid peroxidation and alterations in erythrocyte antioxidant enzyme activities in diabetic patients (Arai et al., 1987; Sharma et al., 2000) as observed in heart, pancreas and blood of STZ induced diabetic rats (Kakkar et al., 1995). In another instance, TBARS is considered as an indicator of free radical production. An increase in TBARS level in liver may therefore be due to raised level of oxidative stress that might promote DNA and protein alterations (Wolff et al., 1991), including; changes in the enzyme activities implicated in lipid metabolism and free radicals scavenging process (Douillet et al., 1998).
Raised level of oxidative stress accounts for low erythrocytes count because of low levels of erythrocyte GSH coupled with increased utilization of GSH, in efforts to ameliorate oxidative stress associated with diabetic erythrocytes (Jain and McVie, 1994). Consequently, pathophysiology of DM promotes oxidative damages of phospholipids and associated biomolecules of erythrocyte membrane. This is supported by the fact that erythrocytes of diabetic patients are more susceptible to lipid peroxidation when treated with H2O2 in vitro (Matkovics et al., 1982; Uzel et al., 1987). In addition, low hematocrit (PCV) percentage may be attributed to the reduction in the total red blood cell count due to failure in blood osmoregulation and elevation of plasma osmolarity (Evan-Wong and Davidson, 1983).
Diabetes mellitus induced alterations in antioxidant enzymes activities: Several studies on tissue levels of activity of enzymatic antioxidant systems are characterized with divergent results. For instance, studies using STZ-treated diabetic rats close to three decades ago showed that increase in pancreatic superoxide dismutase (SOD) activity might be an adaptive response to low pancreatic SOD level, whereas reduction in SOD activity in liver and kidney has direct linkage with the damaging effect of free radicals on the enzyme (Wohaieb and Godin, 1987). In another report, Pieper et al. (1995) demonstrated that in experimental DM, the activity of CAT was elevated in vascular tissues, whereas no significant alterations in the activity of other major antioxidant enzymes {SOD and glutathione peroxidase (GSH-Px)} were noted. Ojiako et al. (2015) reported that levels of renal and hepatocyte antioxidant enzymes (GPOx, SOD, CAT) and low molecular weight antioxidant (LMWA) (GSH/GSSG ratio) were altered in alloxan-induced hyperglycemic rats. In addition, Wohaieb and Godin (1987) reported increased CAT and SOD activities in pancreatic tissues of DM rats, whereas the hepatocytes showed generalized low CAT, SOD and GSH-Px activities. They noted that increase in both CAT and SOD activities occurred in tissues with the lowest antioxidant enzymatic activities (pancreas) before onset of DM. Thus, suggesting a compensatory response to an increase in endogenous oxidant radicals in the pancreas of DM rats. Decreased tissue concentrations of antioxidants, such as vitamin E, SOD and CAT, have also been demonstrated in vitro (Wohaieb and Godin, 1987).
Low levels of GSH in erythrocytes of DM subjects is as a result of low activities of the enzymes involved in GSH synthesis (γ-glutamylcystein synthetase) and/or in the export of oxidized glutathione (GSSG) out of the cell (Murakami et al., 1989) as well as enhanced sorbitol pathway (Ciuchi et al., 1996). In addition, low activity of GSSG-R, which acts to reduce GSSG to GSH, has also been reported in DM (Tagami et al., 1992). Murakami et al. (1989) and Matkovics et al. (1998) reported low level of activity of GSSG-R in erythrocyte haemolysate of STZ-induced DM rats, which they attributed to be the effect of enzyme glycation in uncontrolled hyperglycemia (Jain and McVie, 1994). Also, earlier reports showed significant reduction in the level of activity of erythrocyte GSH-Px in diabetic children and adolescents when compared with that of the control subjects (Dominguez et al., 1998). These previous reports attributed low level of activity of erythrocyte GSH-Px to low blood GSH content in DM subjects, since GSH is a substrate and cofactor for GSH-Px activity. Therefore, low GSH content resulted in corresponding low GSH-Px activity and propensity to elicit oxidative stress. Accordingly, enzyme inactivation either through glycation process (Arai et al., 1987) or under conditions of increased oxidative stress also contribute to low GSH-Px activity (Lyons, 1991).
Antioxidant defenses mechanisms are often impaired in DM with corresponding hyperoxidative stress (Rolo and Palmeira, 2006; Bajaj and Khan, 2012). Furthermore, there is evidence to suggest that DM induces alterations in the activities of antioxidant enzymes in various tissues (Oberley, 1988; Ojiako et al., 2015). Theoretically, alterations in antioxidant enzyme activity are consequences of oxidative stress, glycation of antioxidant enzymes/proteins and disturbances in micronutrient status in DM (Szaleczky et al., 1999; Kang, 2003; Yuan et al., 2010).
CONCLUSION
The critical roles of overwhelming cellular levels of RONS play in the pathophysiology of DM have been incontrovertibly established. Intuitively, the inhibition and/or scavenging of intracellular free radical formation provide a therapeutic strategy to ameliorate oxidative stress and prevent ensuing pathologic complications associated with DM. Therefore, the integration of antioxidants formulations into conventional therapeutic interventions, both by ingestion of natural antioxidants or through dietary supplementation, should be encouraged for a holistic approach to the management and prevention of DM and associated complications. However, despite the obvious usefulness and potential merit/advantages of antioxidant pharmacotherapy, there is still the need to investigate and evaluate the efficacy and safety scores of this therapeutic strategy. Moreover, previous studies on the effect of certain LMWAs on endothelial dysfunction in T2DM revealed contradictory results. Besides, the query of whether antioxidants could have beneficial effect by reducing the risks associated with DM, especially, cardiovascular disease has remained unresolved and inconclusive.
Finally, another novel approach to DM therapy is to provoke over-expression of antioxidant enzymes in a tissue-specific manner, as exemplified in genetic mutant mice model, to serve as control measure against the development of metabolic diseases associated with oxidative stress. This proposed DM therapy shared similar concepts with the reports of Alfadda and Sallam (2012) in which they noted that activation of transcription nuclear factor, nuclear factor-erythroid 2-related factor 2 (Nrf2) induced several antioxidant and detoxification genes in patients with lung cancer. Unfortunately, the metabolic fallouts and effect of this proposed therapeutic approach on general haemostasis of DM individuals is yet to be elucidated.
REFERENCES
- Ahmed, R.G., 2005. The physiological and biochemical effects of diabetes on the balance between oxidative stress and antioxidant defense system. Med. J. Islamic World Acad. Sci., 15: 31-42.
Direct Link - Alfadda, A.A. and R.M. Sallam, 2012. Reactive oxygen species in health and disease. J. Biomed. Biotechnol.
CrossRefDirect Link - Alipour, M., I. Salehi and F.G. Soufi, 2012. Effect of exercise on diabetes-induced oxidative stress in the rat hippocampus. Iran. Red. Cresc. Med. J., 14: 222-228.
Direct Link - Arai, K., S. Maguchi, S. Fujii, H. Ishibashi, K. Oikawa and N. Taniguchi, 1987. Glycation and inactivation of human Cu-Zn-superoxide dismutase. Identification of the in vitro glycated sites. J. Biol. Chem., 262: 16969-16972.
CrossRefPubMedDirect Link - Aronson, D. and E.J. Rayfield, 2002. How hyperglycemia promotes atherosclerosis: Molecular mechanisms. Cardiovasc. Diabetol., Vol. 1.
CrossRefDirect Link - Ashokkumar, N., L. Pari and K.M. Ramkumar, 2006. N-Benzoyl-D-phenylalanine attenuates brain acetylcholinesterase in neonatal streptozotocin-diabetic rats. Basic Clin. Pharmacol. Toxicol., 99: 246-250.
CrossRefPubMedDirect Link - Aydin, A., H. Orhan, A. Sayal, M. Ozata, D. Sahin and A. Isimer, 2001. Oxidative stress and nitric oxide related parameters in type II diabetes mellitus: Effects of glycemic control. Clin. Biochem., 34: 65-70.
Direct Link - Bajaj, S. and A. Khan, 2012. Antioxidants and diabetes. Indian J. Endocrinol. Metab., 16: S267-S271.
CrossRefDirect Link - Basta, G., A.M. Schmidt and R. de Caterina, 2004. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res., 63: 582-592.
PubMedDirect Link - Beckman, J.S., T.W. Beckman, J. Chen, P.A. Marshall and B.A. Freeman, 1990. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA., 87: 1620-1624.
CrossRefPubMedDirect Link - Bierhaus, A., S. Chevion, M. Chevion, M. Hofmann and P. Quehenberger et al., 1997. Advanced glycation end product-induced activation of NF-κB is suppressed by α-lipoic acid in cultured endothelial cells. Diabetes, 46: 1481-1490.
CrossRefDirect Link - Boullier, A., D.A. Bird, M.K. Chang, E.A. Dennis and P. Friedman et al., 2001. Scavenger receptors, oxidized LDL and atherosclerosis. Ann. N. Y. Acad. Sci., 947: 214-223.
CrossRefDirect Link - Brownlee, M., 2001. Biochemistry and molecular cell biology of diabetic complications. Nature, 414: 813-820.
CrossRefPubMedDirect Link - Brownlee, M., A. Cerami and H. Vlassara, 1988. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. New Engl. J. Med., 318: 1315-1321.
PubMedDirect Link - Cadenas, E. and K.J.A. Davies, 2000. Mitochondrial free radical generation, oxidative stress and aging. Free Radic. Biol. Med., 29: 222-230.
CrossRefPubMedDirect Link - Cameron, N.E., M.A. Cotter, V. Archibald, K.C. Dines and E.K. Maxfield, 1994. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia, 37: 449-459.
CrossRefDirect Link - Cameron, N.E., M.A. Cotter, M. Basso and T.C. Hohman, 1997. Comparison of the effects of inhibitors of aldose reductase and sorbitol dehydrogenase on neurovascular function, nerve conduction and tissue polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia, 40: 271-281.
CrossRefDirect Link - Ceolotto, G., M. Bevilacqua, I. Papparella, E. Baritono and L. Franco et al., 2004. Insulin generates free radicals by an NAD(P)H, phosphatidylinositol 3'-kinase-dependent mechanism in human skin fibroblasts ex vivo. Diabetes, 53: 1344-1351.
CrossRefDirect Link - Ceriello, A., 2000. Oxidative stress and glycemic regulation. Metabolism, 49: 27-29.
CrossRefPubMedDirect Link - Ceriello, A., A. Quatraro and D. Giugliano, 1992. New insights on non-enzymatic glycosylation may lead to therapeutic approaches for the prevention of diabetic complications. Diabetic Med., 9: 297-299.
CrossRefDirect Link - Ceriello, A., 1995. Is oxidative stress the missing link between insulin resistance and atherosclerosis? Diabetologia, 38: 1484-1485.
CrossRefDirect Link - Ceriello, A., P.D. Russo, P. Amstad and P. Cerutti, 1996. High glucose induces antioxidant enzymes in human endothelial cells in culture: Evidence linking hyperglycemia and oxidative stress. Diabetes, 45: 471-477.
CrossRefDirect Link - Chikezie, P.C. and A.A. Uwakwe, 2014. Activities of three erythrocyte enzymes of hyperglycemic rats (Rattus norvegicus) treated with Allium sativa extract. J. Diabetes Metab. Disord., Vol. 13.
Direct Link - Chis, I.C., M.I. Ungureanu, A. Marton, R. Simedrea, A. Muresan, I.D. Postescu and N. Decea, 2009. Antioxidant effects of a grape seed extract in a rat model of diabetes mellitus. Diab. Vasc. Dis. Res., 6: 200-204.
CrossRefPubMedDirect Link - Cimato, A.N., G.B. Facorro, L.L. Piehl, M.M.M. Sarrasague, D. Grinspon, H.A. Farach and E.R. de Celis, 2008. Oxidative damage and antioxidant status in diabetes mellitus and rheumatoid arthritis: A comparative study. Open Clin. Chem. J., 1: 92-98.
CrossRefDirect Link - Ciuchi, E., P. Odetti and R. Prando, 1996. Relationship between glutathione and sorbitol concentrations in erythrocytes from diabetic patients. Metabolism, 45: 611-613.
CrossRefDirect Link - Cohen, R.A., 1993. Dysfunction of vascular endothelium in diabetes mellitus. Monogr. Am. Heart Assoc., 87: V67-V76.
Direct Link - Cumaoglu, A., C. Cevik, L. Rackova, N. Ari and C. Karasu, 2007. Effects of antioxidant stobadine on protein carbonylation, advanced oxidation protein products and reductive capacity of liver in streptozotocin-diabetic rats: Role of oxidative/nitrosative stress. BioFactors, 30: 171-178.
CrossRefPubMedDirect Link - Dallak, M.M., D.P. Mikhailidis, M.A. Haidara, I.M. Bin-Jaliah and O.M. Tork et al., 2008. Oxidative stress as a common mediator for apoptosis induced-cardiac damage in diabetic rats. Open Cardiovasc. Med. J., 2: 70-78.
CrossRefDirect Link - Dandona, P., K. Thusu, S. Cook, B. Snyder, J. Makowski, D. Armstrong and T. Nicotera, 1996. Oxidative damage to DNA in diabetes mellitus. Lancet, 347: 444-445.
CrossRefPubMedDirect Link - Dandona, P., P. Mohanty, H. Ghanim, A. Aljada and R. Browne et al., 2001. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation and protein carbonylation. J. Clin. Endocrinol. Metab., 86: 355-362.
CrossRefPubMedDirect Link - Davison, G.W., L. George, S.K. Jackson, I.S. Young and B. Davies et al., 2002. Exercise, free radicals and lipid peroxidation in type 1 diabetes mellitus. Free Radic. Biol. Med., 33: 1543-1551.
CrossRefDirect Link - DeFronzo, R.A. and E. Ferrannini, 1991. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care, 14: 173-194.
PubMedDirect Link - Desco, M.C., M. Asensi, R. Marquez, J. Martinez-Valls and M. Vento et al., 2002. Xanthine oxidase is involved in free radical production in type 1 diabetes protection by Allopurinol. Diabetes, 51: 1118-1124.
CrossRefDirect Link - Dominguez, C., E. Ruiz, M. Gussinye and A. Carrascosa, 1998. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care, 21: 1736-1742.
CrossRefDirect Link - Douillet, C., M. Bost, M. Accominotti, F. Borson-Chazot and M. Ciavatti, 1998. Effect of selenium and vitamin E supplements on tissue lipids, peroxides and fatty acid distribution in experimental diabetes. Lipids, 33: 393-399.
CrossRefDirect Link - El-Missiry, M.A., A.I. Othman and M.A. Amer, 2004. L-Arginine ameliorates oxidative stress in alloxan-induced experimental diabetes mellitus. J. Applied Toxicol., 24: 93-97.
CrossRefDirect Link - El-Wassef, M., G.S.M. El-Saeed, S.E. El-Tokhy, H.M. Raslan, S. Tawfeek, I. Siam and S.I. Salem, 2012. Oxidative DNA damage in patients with type 2 diabetes mellitus. Diabetologia Croatica, 41: 121-127.
Direct Link - Esposito, K., F. Nappo, R. Marfella, G. Giugliano and F. Giugliano et al., 2002. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation, 106: 2067-2072.
CrossRefDirect Link - Evans, J.L., I.D., Goldfine, B.A. Maddux and G.M. Grodsky, 2003. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes, 52: 1-8.
CrossRefPubMedDirect Link - Feener, E.P. and G.L. King, 1997. Vascular dysfunction in diabetes mellitus. Lancet, 350: S9-S13.
PubMedDirect Link - Ghareeb, D.A. and H.M. Hussen, 2008. Vanadium improves brain acetylcholinesterase activity on early stage alloxan-diabetic rats. Neurosci. Lett., 436: 44-47.
CrossRefDirect Link - Giacco, F., M. Brownlee and A.M. Schmidt, 2010. Oxidative stress and diabetic complications. Circ. Res., 107: 1058-1070.
CrossRefPubMedDirect Link - Golbidi, S., M. Badran and I. Laher, 2012. Antioxidant and anti-inflammatory effects of exercise in diabetic patients. Exp. Diabetes Res., Vol. 2012.
Direct Link - Griffin, M.E., D. Mclnerney, A. Fraser, A.H. Johnson, P.B. Collins and G.H. Owens, 1997. Autoantibodies to oxidized low density lipoprotein: The relationship to low density lipoprotein fatty acid composition in diabetes. Diabetes Med., 14: 741-747.
CrossRefDirect Link - Grimsrud, P.A., H. Xie, T.J. Griffin and D.A. Bernlohr, 2008. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem., 283: 21837-21841.
CrossRefDirect Link - Habib, M.P., F.D. Dickerson and A.D. Mooradian, 1994. Effect of diabetes, insulin and glucose load on lipid peroxidation in the rat. Metabolism, 43: 1442-1445.
CrossRefDirect Link - Hammes, H.P., A. Bartmann, L. Engi and P. Wulforth, 1997. Antioxidant treatment of experimental diabetic retinopathy in rats with nicanartine. Diabetologia, 40: 629-634.
CrossRefDirect Link - Hartog, J.W.L., A.A. Voors, S.J.L. Bakker, A.J. Smit and D.J. van Veldhuisen, 2007. Advanced glycation end-products (AGEs) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail., 9: 1146-1155.
CrossRefDirect Link - Hayden, M.R. and S.C. Tyagi, 2004. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J., Vol. 3.
Direct Link - Higuchi, Y., 1982. Lipid peroxides and alpha-tocopherol in rat streptozotocin-induced diabetes mellitus. Acta. Med. Okayama., 36: 165-175.
PubMedDirect Link - Ihara, Y., K. Egashira, K. Nakano, K. Ohtani and M. Kubo et al., 2007. Upregulation of the ligand-RAGE pathway via the angiotensin II type I receptor is essential in the pathogenesis of diabetic atherosclerosis. J. Mol. Cell. Cardiol., 43: 455-464.
CrossRefDirect Link - Inoguchi, T., T. Sonta, H. Tsubouchi, T. Etoh and M. Kakimoto et al., 2003. Protein kinase C-dependent increase in Reactive Oxygen Species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol., 14: S227-S232.
PubMedDirect Link - Jaganjac, M., O. Tirosh, G. Cohen, S. Sasson and N. Zarkovic, 2013. Reactive aldehydes-second messengers of free radicals in diabetes mellitus. Free Radic. Res., 47: 39-48.
CrossRefDirect Link - Jain, S.K. and R. McVie, 1994. Effect of glycemic control, race (white versus black), and duration of diabetes on reduced glutathione content in erythrocytes of diabetic patients. Metabolism, 43: 306-309.
CrossRefDirect Link - Jain, S.K., R. McVie, J.J. Jaramillo and Y. Chen, 1998. Hyperketonemia (Acetoacetate) increases the oxidizability of LDL + VLDL in type-I diabetic patients. Free Rad. Biol. Med., 24: 175-181.
CrossRefDirect Link - Jorns, A., M. Tiedge, S. Lenzen and R. Munday, 1999. Effect of superoxide dismutase, catalase, chelating agents and free radical scavengers on the toxicity of alloxan to isolated pancreatic islets in vitro. Free Radic. Biol. Med., 26: 1300-1304.
CrossRefDirect Link - Kajanachumpol, S., S. Komindr and A. Mahaisiriyodom, 1997. Plasma lipid peroxide and antioxidant levels in diabetic patients. J. Med. Assoc. Thai., 80: 372-377.
PubMedDirect Link - Kakkar, R., J. Kalra, S.V. Mantha and K. Prasad, 1995. Lipid peroxidation and activity of antioxidant enzymes in diabetic rats. Mol. Cell. Biochem., 151: 113-119.
CrossRefDirect Link - Kaneki, M., N. Shimizu, D. Yamada and K. Chang, 2007. Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox. Signal., 9: 319-329.
Direct Link - Kang, J.H., 2003. Modification and inactivation of human Cu, Zn-superoxide dismutase by methylglyoxal. Mol. Cells, 15: 194-199.
PubMedDirect Link - Murakami, K., T. Kondo, Y. Ohtsuka, Y. Fujiwara, M. Shimada and Y. Kawakami, 1989. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism, 38: 753-758.
PubMedDirect Link - Keaney, Jr. J.F. and J. Loscalzo, 1999. Diabetes, oxidative stress and platelet activation. Circulation, 99: 189-191.
CrossRefDirect Link - Kobayashi, K. and J.R. Neely, 1979. Control of maximum rates of glycolysis in rat cardiac muscle. Circ. Res., 44: 166-175.
PubMedDirect Link - Krieger-Brauer, H.I. and H. Kather, 1992. Human fat cells possess a plasma membrane-bound H2O2-generating system that is activated by insulin via a mechanism bypassing the receptor kinase. J. Clin. Invest., 89: 1006-1013.
CrossRefDirect Link - Landmesser, U., S. Dikalov, S.R. Price, L. McCann and T. Fukai et al., 2003. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest., 111: 1201-1209.
CrossRefDirect Link - Lassila, M., K.K. Seah, T.J. Allen, V. Thallas and M.C. Thomas et al., 2004. Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: Role of advanced glycation end products. J. Am. Soc. Nephrol., 15: 2125-2138.
PubMedDirect Link - Lee, T.S., K.A. Saltsman, H. Ohashi and G.L. King, 1989. Activation of protein kinase C by elevation of glucose concentration: Proposal for a mechanism in the development of diabetic vascular complications. Proc. Natl. Acad. Sci. USA., 86: 5141-5145.
Direct Link - Lehto, S., T. Ronnemaa, S.M. Haffner, K. Pyorala, V. Kallio and M. Laakso, 1997. Dyslipidemia and hyperglycemia predict coronary heart disease events in middle-aged patients with NIDDM. Diabetes, 46: 1354-1359.
PubMedDirect Link - Levine, R.L., 1983. Oxidative modification of glutamine synthetase. I. Inactivation is due to loss of one histidine residue. J. Biol. Chem., 258: 11823-11827.
Direct Link - Levine, R.L., B.S. Berlett, J. Moskovitz, L. Mosoni and E.R. Stadtman, 1999. Methionine residues may protect proteins from critical oxidative damage. Mech. Ageing Dev., 107: 323-332.
CrossRefDirect Link - Lyons, T.J., 1991. Oxidized low density lipoproteins: A role in the pathogenesis of atherosclerosis in diabetes? Diabetes Med., 8: 411-419.
CrossRefDirect Link - Maejima, K., S. Nakano, M. Himeno, S.I. Tsuda and H. Makiishi et al., 2001. Increased basal levels of plasma nitric oxide in type 2 diabetic subjects: Relationship to microvascular complications. J. Diabetes Complications, 15: 135-143.
CrossRefDirect Link - Matkovics, B., M. Kotorman, I.S. Varga, D.Q. Hai and C. Varga, 1998. Oxidative stress in experimental diabetes induced by streptozotocin. Acta Physiologica Hungarica, 85: 29-38.
PubMedDirect Link - Matkovics, B., S.I. Varga, L. Szabo and H. Witas, 1982. The effect of diabetes on the activities of the peroxide metabolism enzymes. Hormone Metab. Res., 14: 77-79.
CrossRefPubMedDirect Link - Matteucci, E. and O. Giampietro, 2000. Oxidative stress in families of type 1 diabetic patients. Diabetes Care, 23: 1182-1186.
PubMedDirect Link - Monnier, V.M., 1990. Nonenzymatic glycosylation, the Maillard reaction and the aging process. J. Gerontol., 45: B105-B111.
CrossRefPubMedDirect Link - Moussa, S.A., 2008. Oxidative stress in diabetes mellitus. Rom. J. Biophys., 18: 225-236.
Direct Link - Muellenbach, E.A., C.J. Diehl, M.K. Teachey, K.A. Lindborg and T.L. Archuleta et al., 2008. Interactions of the advanced glycation end product inhibitor pyridoxamine and the antioxidant α-lipoic acid on insulin resistance in the obese Zucker rat. Metabolism, 57: 1465-1472.
CrossRefDirect Link - Musabayane, C.T., 2012. The effects of medicinal plants on renal function and blood pressure in diabetes mellitus. Cardiovasc. J. Afr., 23: 462-468.
PubMedDirect Link - Nishigaki, J., M. Hagihara, H. Tsunekawa, M. Maseki and K. Yagi, 1981. Lipid peroxide levels of serum lipoprotein fractions of diabetic patients. Biochem. Med., 25: 373-378.
PubMedDirect Link - Oberley, L.W., 1988. Free radicals and diabetes. Free Radical Biol. Med., 5: 113-124.
CrossRefDirect Link - Ojiako, A.O., P.C. Chikezie and C.A. Ogbuji, 2015. Renal and hepatic antioxidant status of hyperglycemic rats treated with single and combinatorial herbal formulations. Pharmacogn. Commun., 5: 148-159.
CrossRefDirect Link - Packer, L., 1993. The role of anti-oxidative treatment in diabetes mellitus. Diabetologia, 36: 1212-1213.
CrossRefDirect Link - Pautler, E.L. and S.R. Ennis, 1980. The effect of induced diabetes on the electroretinogram components of the pigmented rat. Invest. Ophthalmol. Visual Sci., 19: 702-705.
PubMedDirect Link - Perez-Matute, P., M.A. Zulet and J.A. Martinez, 2009. Reactive species and diabetes: Counteracting oxidative stress to improve health. Curr. Opin. Pharmacol., 9: 771-779.
CrossRefDirect Link - Pierce, A., H. Mirzaei, F. Muller, E. De Waal and A.B. Taylor et al., 2008. GAPDH is conformationally and functionally altered in association with oxidative stress in mouse models of amyotrophic lateral sclerosis. J. Mol. Biol., 382: 1195-1210.
CrossRefDirect Link - Pieper, G.M., M. Jordan, L.A. Dondlinger, M.B. Adams and A.M. Roza, 1995. Peroxidative stress in diabetic blood vessels. Reversal by pancreatic islet transplantation. Diabetes, 44: 884-889.
PubMedDirect Link - Poljsak, B. and R. Fink, 2014. The protective role of antioxidants in the defence against ROS/RNS-mediated environmental pollution. Oxidative Med. Cell. Longevity., 1: 1-22.
CrossRefDirect Link - Pollack, M. and C. Leeuwenburgh, 2000. Molecular Mechanisms of Oxidative Stress in Aging: Free Radicals, Aging, Antioxidants and Disease. In: Handbook of Oxidants and Antioxidants in Exercise, Sen, C.K., L. Packer and O. Hanninen (Eds.). Elsevier Science, Amsterdam, The Netherlands, ISBN-13: 9780080538297, pp: 881-923.
- Poston, L. and P.D. Taylor, 1995. Endothelium-mediated vascular function in insulin-dependent diabetes mellitus. Clin. Sci., 88: 245-255.
Direct Link - Quagliaro, L., L. Piconi, R. Assaloni, L. Martinelli, E. Motz and A. Ceriello, 2003. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes, 52: 2795-2804.
CrossRefDirect Link - Randle, P.J., D.A. Priestman, S. Mistry and A. Halsall, 1994. Mechanisms modifying glucose oxidation in diabetes mellitus. Diabetologia, 37: S155-S161.
CrossRefDirect Link - Rerup, C.C., 1970. Drugs producing diabetes through damage of the insulin secreting cells. Pharmacol. Rev., 22: 485-518.
PubMedDirect Link - Rolo, A.P. and C.M. Palmeira, 2006. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Applied Pharmacol., 212: 167-178.
CrossRefDirect Link - Rotilio, G., M.T. Carr, L. Rossi and M.R. Ciriolo, 2000. Copper-dependent oxidative stress and neurodegeneration. IUBMB Life, 50: 309-314.
CrossRefDirect Link - Rowe, J.W., J.B. Young, K.L. Minaker, A.L. Stevens, J. Pallotta and L. Landsberg, 1981. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes, 30: 219-225.
CrossRefDirect Link - Rungby, J., A. Flyvbjerg, H.B. Andersen and K. Nyborg, 1992. Lipid peroxidation in early experimental diabetes in rats: Effects of diabetes and insulin. Acta Endocrinologica, 126: 378-380.
CrossRefDirect Link - Santilli, F., F. Cipollone, A. Mezzetti and F. Chiarelli, 2004. The role of nitric oxide in the development of diabetic angiopathy. Hormone Metab. Res., 36: 319-335.
CrossRefPubMedDirect Link - Santini, S.A., G. Marra, B. Giardina, P. Controneo and A. Mordente et al., 1997. Defective plasma antioxidant defenses and enhanced susceptibility to lipid peroxidation in uncomplicated IDDM. Diabetes, 46: 1853-1858.
CrossRefDirect Link - Sato, Y., N. Hotta, N. Sakamoto, S. Matsuoks, N. Ohishi and K. Yagi, 1979. Lipid peroxide level in plasma of diabetic patients. Biochem. Med., 21: 104-107.
CrossRefDirect Link - Schiekofer, S., M. Andrassy, J. Chen, G. Rudofsky and J. Schneider et al., 2003. Acute hyperglycemia causes intracellular formation of CML and activation of ras, p42/44 MAPK and nuclear factor κB in PBMCs. Diabetes, 52: 621-633.
CrossRefDirect Link - Sellamuthu, P.S., P. Arulselvan, S. Kamalraj, S. Fakurazi and M. Kandasamy, 2013. Protective nature of mangiferin on oxidative stress and antioxidant status in tissues of streptozotocin-induced diabetic rats. ISRN Pharmacol.
CrossRefDirect Link - Sharma, A., S. Kharb, S.N. Chug, R. Kakkar and G.P. Singh, 2000. Evaluation of oxidative stress before and after control of glycemia and after vitamin E supplementation in diabetic patients. Metabolism, 49: 160-162.
CrossRefDirect Link - Shin, C.S., B.S. Moon, K.S. Park, S.Y. Kim, S.J. Park, M.H. Chung and H.K. Lee, 2001. Serum 8-hydroxy-guanine levels are increased in diabetic patients. Diabetes Care, 24: 733-737.
CrossRefDirect Link - Singal, P.K., R.E. Beamish and N.S. Dhalla, 1983. Potential oxidative pathways of catecholamines in the formation of lipid peroxides and genesis of heart disease. Adv. Exp. Med. Biol., 161: 391-401.
CrossRefDirect Link - Skrha, J., A. Hodinar, J. Kvasnicka and J. Hilgertova, 1996. Relationship of oxidative stress and fibrinolysis in diabetes mellitus. Diabetic Med., 13: 800-805.
CrossRefDirect Link - Spitaler, M.M. and W.F. Graier, 2002. Vascular targets of redox signalling in diabetes mellitus. Diabetologia, 45: 476-494.
CrossRefDirect Link - Styskal, J., H. van Remmen, A. Richardson and A.B. Salmon, 2012. Oxidative stress and diabetes: What can we learn about insulin resistance from antioxidant mutant mouse models? Free Radical Biol. Med., 52: 46-58.
CrossRefDirect Link - Sugatani, J., T. Wada, M. Osabe, K. Yamakawa, K. Yoshinari and M. Miwa, 2006. Dietary inulin alleviates hepatic steatosis and xenobiotics-induced liver injury in rats fed a high-fat and high-sucrose diet: Association with the suppression of hepatic cytochrome P450 and hepatocyte nuclear factor 4 alpha expression. Drug Metabol. Disposition., 34: 1677-1687.
PubMedDirect Link - Szaleczky, E., J. Prechl, J. Feher and A. Somogyi, 1999. Alterations in enzymatic antioxidant defence in diabetes mellitus-a rational approach. Postgrad. Med. J., 75: 13-17.
CrossRefDirect Link - Tagami, S., T. Kondo, K. Yoshida, J. Hirokawa, Y. Ohtsuka and Y. Kawakami, 1992. Effect of insulin on impaired antioxidant activities in aortic endothelial cells from diabetic rabbits. Metabolism, 41: 1053-1058.
CrossRefDirect Link - Tesfamariam, B. and R.A. Cohen, 1992. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am. J. Physiol., 263: 321-326.
PubMedDirect Link - Thannickal, V.J. and B.L. Fanburg, 2000. Reactive oxygen species in cell signaling. Am. J. Physiol.-Lung Cell. Mol. Physiol., 279: L1005-L1028.
CrossRefDirect Link - Tilton, R.G., K. Chang, J.R. Nyengaard, M. van den Enden, Y. Ido and J.R. Williamson, 1995. Inhibition of sorbitol dehydrogenase: Effects on vascular and neural dysfunction in streptozocin-induced diabetic rats. Diabetes, 44: 234-242.
CrossRefDirect Link - Toborek, M. and B. Hennig, 1994. Fatty acid-mediated effects on the glutathione redox cycle in cultured endothelial cells. Am. J. Clin. Nutr., 59: 60-65.
Direct Link - Toborek, M., T. Wasik, M. Drozdz, M. Klin, K. Magner-Wrobel and E. Kopieczna-Grzebieniak, 1992. Effect of hemodialysis on lipid peroxidation and antioxidant system in patients with chronic renal failure. Metabolism, 41: 1229-1232.
CrossRefDirect Link - Uzel, N., A. Sivas, M. Uysal and H. Oz, 1987. Erythrocyte lipid peroxidation and glutathione peroxidase activities in patients with diabetes mellitus. Hormone Metab. Res., 19: 89-90.
CrossRefPubMedDirect Link - Vaag, A., P. Damsbo, O. Hother-Nielsen and H. Beck-Nielsen, 1992. Hyperglycaemia compensates for the defects in insulin-mediated glucose metabolism and in the activation of glycogen synthase in the skeletal muscle of patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia, 35: 80-88.
CrossRefDirect Link - Violi, F., R. Marino, M.T. Milite and L. Loffredo, 1999. Nitric oxide and its role in lipid peroxidation. Diabetes/Metab. Res. Rev., 15: 283-288.
CrossRefDirect Link - Williamson, J.R., K. Chang, M. Frangos, K.S. Hasan and Y. Ido et al., 1993. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes, 42: 801-813.
PubMedDirect Link - Wohaieb, S.A. and D.V. Godin, 1987. Alterations in free radical tissue-defense mechanisms in Streptozocin-induced diabetes in rat: Effects of insulin treatment. Diabetes, 36: 1014-1018.
CrossRefDirect Link - Wolf, G. and F.N. Ziyadeh, 2007. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol., 106: 26-31.
CrossRefPubMedDirect Link - Wolff, S.P., 1993. Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br. Med. Bull., 49: 642-652.
PubMedDirect Link - Wolff, S.P., Z.Y. Jiang and J.V. Hunt, 1991. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Rad. Biol. Med., 10: 339-352.
PubMedDirect Link - Evan-Wong, L.A. and R.J. Davidson, 1983. Raised Coulter mean corpuscular volume in diabetic ketoacidosis and its underlying association with marked plasma hyperosmolarity. J. Clin. Pathol., 36: 334-336.
CrossRefPubMedDirect Link - Wright, Jr.E., J.L. Scism-Bacon and L.C. Glass, 2006. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract., 60: 308-314.
CrossRefDirect Link - Yonekura, H., Y. Yamamoto, S. Sakurai, T. Watanabe and H. Yamamoto, 2005. Roles of the receptor for advanced glycation endproducts in diabetes-induced vascular injury. J. Pharmacol. Sci., 97: 305-311.
CrossRefDirect Link - Yuan, Y., X. Jiao, W.B. Lau, Y. Wang and T.A. Christopher et al., 2010. Thioredoxin glycation: A novel posttranslational modification that inhibits its antioxidant and organ protective actions. Free Radical Biol. Med., 49: 332-338.
CrossRefDirect Link - Yung, L.M., F.P. Leung, X. Yao, Z.Y. Chen and Y. Huang, 2006. Reactive oxygen species in vascular wall. Cardiovasc. Hematol. Disord. Drug Targets, 6: 1-19.
PubMedDirect Link