
Abu Sadeque Md. Selim
Bangladesh Open University, Bangladesh







ABSTRACT
Molecular techniques those have been applied for analyzing chicken microbiota have been summarized here. Since, the knowledge of molecular analysis of chicken microbiota as well as intestinal ecosystem is still limited, this review will encourage animal scientists/microbiologists to apply various types of molecular techniques for monitoring intestinal microbes.
PDF Abstract XML References
How to cite this article
Abu Sadeque Md. Selim, 2006. Molecular Techniques for Analyzing Chicken Microbiota. Biotechnology, 5: 53-57.
DOI: 10.3923/biotech.2006.53.57
URL: https://scialert.net/abstract/?doi=biotech.2006.53.57
DOI: 10.3923/biotech.2006.53.57
URL: https://scialert.net/abstract/?doi=biotech.2006.53.57
INTRODUCTION
The normal microbiota of the gastro-intestinal tract works as a barrier against pathogens, contributes to degradation of some food components, stimulates the host immune system and produces enzymes and short-chain fatty acids (Stiles and Holzapfel, 1997). The microbes can also metabolize potentially carcinogenic substances and drugs in either a beneficial or a disadvantageous way (Stiles and Holzapfel, 1997). However, the role and action of individual microbial species or groups present in the gastro-intestinal tract are poorly known.
Recent advances in rRNA-based molecular techniques make it possible to identify different bacterial populations in environmental samples without prior cultivation. In the past most studies with a focus on chicken microbial analysis have been conducted using conventional methods. A small number of previous studies on chicken microbiota have been done applying molecular techniques. Thus the knowledge on available molecular techniques, their application and mechanism to animal scientists for chicken microbiota analysis is poor. This review will attempt to summarize all molecular techniques with their application in chicken microbiota for a better understanding of microbial diversity and its role in the chicken ecosystem maintenance.
Utilization of methods independent of prior knowledge on sequence data.
16S rDNA clone libraries: Cloning and sequencing of the 16S ribosomal DNA (rDNA) pools in a population sample provides a method for obtaining sequence-level information on uncultivable bacteria abundantly present in various parts of the gastrointestinal tract. For example, the predominance of sequences within the Firmicutes phylum (94%) emphasizes the prominence and role of this group in the structure and function of the cecum bacterial community (Lan et al., 2002). The major groups of Firmicutes phylum were Clostridia and Lactobacilli. Clostridium subcluster XIVa was considered as the most predominant group in chicken cecum with 38% of analyzed cloned sequence. The second major group of the chicken cecal microbiota was the Lactobacillus sp. (24%). The results of the 16S rDNA sequence analysis of retrieved clones from the chicken cecum showed that 65% of total lactobacilli detected belonged to the Lactobacillus acidophilus group and 32% Lactobacillus crispatus. The other important strains was Lactobacillus salivarius (20%). Lactobacillus such as Lactobacillus reuteri, Lactobacillus oris, Lactobacillus fermentum were also detected.
Denaturing Gradient Gel Electrophoresis (DGGE): Denaturing gradient gel electrophoresis (DGGE) has become popular method for analysis of microbial populations present in chicken intestine (Guan et al., 2003). This method allows separation of nucleic acid molecules based on their size and sequential differences. Thus, a population of DNA molecules, such as the 16S ribosomal RNA (rRNA) or PCR-amplified 16S rDNA, can be studied and predominant members of the population can be identified via sequencing of isolated nucleic acid bands. Sensitivity of gradient gel electrophoresis is affected by the choice of PCR primers used. For example, utilization of universal 16S primers limits the sensitivity to detection of 1% subpopulations (Wilson and Blitchington, 1996). By contrast, utilization of species or group-specific primers may allow detection and identification of bacteria representing a minority of the total population (Zoetendal et al., 1998). Sequence analysis of DGGE fragments demonstrated, for the first time, the presence of L. johnsonii and L. crispatus and/or L. gallinarum in the crop of broilers (Guan et al., 2003).
Terminal restriction fragment length polymorphism (T-RFLP): Terminal restriction fragment length polymorphism (T-RFLP) is based on endonuclease digestion of PCR-amplified DNA and capillary electrophoresis analysis of the terminal restriction fragment (TRF) containing a fluorescent label. Terminal restriction patterns have been used to analyze marine bacterioplankton communities (Moeseneder et al., 2001) as well as faecal bacteria (Kaplan et al., 2001). The method is, however, limited by the choice of primers, which can, with their different affinities, dramatically change the patterns observed, while another problem is the TRF length overlap by phylogenetically distant bacteria (Kaplan et al., 2001). The diversity of intestinal bacteria using TRF has been monitored (Gong et al., 2002), the ileal bacteria were found to be less diverse than the cecal. Lactobacilli, E. cecorumand butyrate-producing bacteria were the three major groups detected in ceca and ilea.
Utilization of specific oligonucleotide primers or probes.
Hybridization: Hybridization offers a means for direct semi-quantitative monitoring of population samples. A very high sensitivity for detection of DNA targets can be obtained with radioactively labeled probes (Palva, 1985; Yamamoto et al., 1992). However, because of its abundance in bacterial cells, rRNA provides for a more attractive target for hybridization studies. Indeed, hybridization assays targeting rDNA have been verified as 10-fold less sensitive than assays for rRNA (Muttray and Mohn, 2000). Dot blot hybridization with rRNA targeted probes has been used for semi quantitative analysis of intestinal microbiota (Hopkins et al., 2001).
Principle of fluorescence in situ hybridization (FISH): The basic principle of the method is that single-stranded DNA binds or anneals to its complementary DNA sequence (Fig. 1). Thus, a DNA probe for a specific chromosomal region recognize and hybridize to its complementary sequence on a metaphase chromosome or within an interphase nucleus. Both have to be in single-strand conformation, therefore the DNA probe and the target DNA must be denatured, usually by heating them in a formamide-containing solution. Actually the probe is hybridized to the target DNA under conditions that allow the DNA to reanneal in double-strand form. Added to the hybridization mixture is an excess of repetitive sequence DNA to block non-specific binding of the probe to the target. After hybridization is complete (often after 2-18 h at 46°C), the slides are washed in formamide-saline citrate solutions to remove excess or non-specifically bound probe.
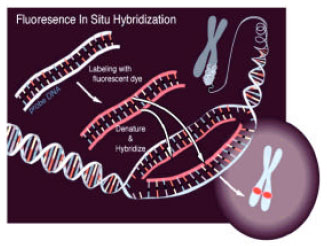 | |
| Fig. 1: | Principle of FISH, Adapted from, National Human Genome Research Institute (NHGRI), by Artist Darryl Leja |
To detect the location of the probe on the target DNA, the probe DNA can be directly labeled with a fluorescent tag. It can also be chemically modified by the addition of hapten molecules (biotin or digoxigenin) that can then be indirectly fluorescently labeled with immunocytochemical techniques. The target DNA is counter stained with another fluorochrome of a complementary color. The probe DNA can be observed on its target by using a fluorescent microscope with filters specific for the fluorochrome label and the counterstain. Special filters have been developed to allow simultaneous visualization of several fluorochromes. Digital cameras designed to detect low light level emissions and computer imaging are used to increase the sensitivity of probe detection. Because fluorescent dyes are subject to photobleaching (fading), the preparations are not permanent and must be stored away from light. Use of an antifade solution (phenylenediamine) has improved the capacity to observe and document fluorescently labeled samples.
Polymerase Chain Reaction (PCR): Amplification of target nucleic acids with PCR is an effortless method for detection of target DNA from various samples. Bacteroides prevotella 16S rDNA markers for human and cows was identified by T-RFLP (Bernhard and Field, 2000; Salanitro et al., 1974) and pathogens from clinical samples by PCR (Harper-Owen et al., 1999). Partial or full amplification of the 16S rDNA by PCR-DGGE or colony hybridization with primers specific for a genus or a few closely related genera has also been reported (Heilig et al., 2002; Kaufmann et al., 1997; Kok et al., 1996; Walter et al., 2001). In addition, utilization of 16S-23S intergenic sequences as targets of species specific primers for different lactic acid bacteria has been described (Tilsala-Timisjärvi and Alatossava, 1997). Good sensitivities have been reported for the PCR detection assays of faecal bacteria. For example, PCR detection sensitivity of five ruminococcal species with species-specific primers from faecal samples spiked with the target species did not markedly differ from the observed detection limit of 4-100 cells from pure cultures (Wang et al., 1997). In spite of a highly potent detection method, results obtained by conventional endpoint PCR should not be considered to be directly quantitative. PCR may lead to differential amplification of target templates originally present in equal amounts (Reysenbach et al., 1992). In addition, very low template concentrations may generate random fluctuations in priming efficiency of population DNA samples with universal primers, leading to a bias in the end-product concentration (Chandler et al., 1997). Knowledge of the rRNA gene copy numbers and genome sizes of bacteria in a mixed DNA sample has been observed to be insufficient in predicting the final product ratio of a PCR amplification. The accumulation of end products during mixed-template PCR caused biasing of the various end-product ratios towards a 1:1 situation, which was hypothesised to be caused by an increase in the homologous template hybridization, decreasing the efficiency of primer annealing and subsequent amplification (Suzuki and Giovannoni, 1996).
Real time PCR: Since the advent of real-time PCR, several techniques have been introduced, including primers with fluorescent dyes, molecular beacons, dual probes and intercalating dyes such as SYBR Green I. Real-time PCR is a superior technique for quantification of nucleic acids. While competitive PCR has been demonstrated to be as reproducible and accurate as real-time PCR, the latter has the benefit of an easier methodology, reducing the need for sample DNA treatment. Although real-time PCR is a relatively new technique, several detection or quantification assays targeting various bacteria have already been described. Target bacteria include carious dentine bacteria (Martin et al., 2002), Desulfotomaculum from soil (Stubner, 2002), faecal bifidobacterial species (Gueimonde et al., 2004; Requena et al., 2002), Campylobacter jejuni (Nogva et al., 2000). Some applications for determination of larger bacterial groups have also been published (Bach et al., 2002; Nadkarni et al., 2002; Suzuki et al., 2000). Real-time PCR is also utilized as a rapid diagnostic tool for detection of pathogenic bacterial species or strains present in a sample. With pathogenic bacteria, the target of choice for real-time PCR is generally a gene associated with the pathogenic traits (Ke et al., 2000; Rauter et al., 2002; Tondella et al., 2002). Detection of PCR amplicons with specific probes is often favored over usage of intercalating dyes due to the better sensitivity and lack of detection of falsely primed products. However, SYBR Green I has become popular because of the possibility to use this intercalating dye in virtually any assay. Real-time quantitative PCR with SYBR Green I has great potential in situations where a diverse target population is to be detected with PCR (Bach et al., 2002). A probe-based methodology requires a binding site for the probe in the vicinity of one of the primers; however, such a conserved site is likely to be missing from a degenerate target DNA population. This should be taken into account, especially when a bacterial population containing several hitherto unknown species is studied.
Real-time PCR is based on on-line measurement of the amplification reaction, enabling quantification of the product during the logarithmic phase of PCR. The first and so far most commonly used real-time PCR approach is the 5´-nuclease (TaqMan) assay introduced. The original assay operates using a radioactively labeled probe that hybridizes in the PCR template region to generate a specific, detectable signal from the amplification reaction. The detection probe becomes annealed to one of the DNA strands during the amplification and is cleaved by the Thermus aquaticus DNA polymerase 5´-exonuclease activity during the primer extension step (Holland et al., 1991). The further extended the 5´-nuclease assay by utilization of doubly labeled oligonucleotide probes for fluorescent measuring of formation of the specific PCR product during primer extension was also reported (Lee et al., 1993).
How Taqman® works: Taqman® utilizes a system that is fairly easy to grasp conceptually. The important thing is to design the probe which consists of two types of fluorophores, the quencher fluorophore (usually a long-wavelength colored dye, such as red) reduces the fluorescence from the reporter fluorophore (usually a short-wavelength colored dye, such as green). It does this by the use of Fluorescence (or Förster) Resonance Energy Transfer (FRET), which is the inhibition of one dye caused by another without emission of a proton. The reporter dye is found on the 5’ end of the probe and the quencher at the 3’ end. When the TaqMan® probe bind to its specific piece of the template DNA after denaturation, the primers anneal to the DNA Taq polymerase and adds nucleotides and remove the Taqman® probe from the template DNA. This separates the quencher from the reporter and allows the reporter to give off its emit its energy. This is then quantified using a computer. The more times the denaturing and annealing takes place, the more opportunities there are for the Taqman® probe to bind and, in turn, the more emitted light is detected.
In conclusion, molecular biology tools are needed for better understanding of the composition and functions of the chicken intestinal microbiota. The successful application of molecular techniques may be a powerful tool to study the distribution and abundance of chicken microbiota in a complex microbial community.
REFERENCES
- Bernhard, A.E. and K.G. Field, 2000. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-prevotella genes encoding 16S rRNA. Applied Environ. Microbiol., 66: 4571-4574.
CrossRefDirect Link - Bach, H.J., J. Tomanova, M. Schloter and J.C. Munch, 2002. Enumeration of total bacteria and bacteria with genes for proteolytic activity in pure cultures and in 45 environmental samples by quantitative PCR mediated amplification. J. Microbiol. Methods, 49: 235-245.
Direct Link - Chandler, D.P., J.K. Fredrickson and F.J. Brockman, 1997. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol. Ecol., 6: 475-482.
Direct Link - Gong, J., R.J. Forster, H. Yu, J.R. Chambers, P.M. Sabour, R. Wheatcroft and S. Chen, 2002. Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS Microbiol. Lett., 208: 1-7.
CrossRefDirect Link - Guan, L.L., K.E. Hagen, G.W. Tannock, D.R. Korver, G.M. Fasenko and G.E. Allison, 2003. Detection and identification of Lactobacillus species in crops of broilers of different ages by using PCR-denaturing gradient gel electro phoresis and amplified Ribosomal DNA restriction analysis. Applied Environ. Microbiol., 69: 6750-6757.
- Gueimonde, M., S. Tolkko, T. Korpimaki and S. Salminen, 2004. New real-time quantitative PCR procedure for quantification of bifidobacteria in human fecal samples. Applied Environ. Microbiol., 70: 4165-4169.
Direct Link - Holland, P.M., R.D. Abramson, R. Watson and D.H. Gelwand, 1991. Detection of specific polymerase chain reaction product by utilizing the 5'-3' exonucleaseactivity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA., 88: 7276-7280.
Direct Link - Holzapfel, W.H., P. Haberer, J. Snel, U. Schillinger and J.H.J.H. Veld, 1998. Overview of gut flora and probiotics. Int. J. Food Microbiol., 41: 85-101.
CrossRefPubMedDirect Link - Haper, O.R., D. Dymock, V. Booth, A.J. Weightman and W.G. Wade, 1999. Detection of unculturable bacteria in periodontal health and disease by PCR. J. Clin. Microbiol., 37: 1469-1473.
PubMed - Hopkins, M.J., R. Sharp and G.T. Macfarlane, 2001. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundanceand community cellular fatty acid profiles. Gut, 48: 198-205.
CrossRefDirect Link - Heilig, H.G.H.J., E.G. Zoetendal, E.E. Vaughan, P. Marteau, A.D.L. Akkermans and W.M. de Vos, 2002. Molecular diversity of Lactobacillus spp. and other lactic acid bacteria in the human intestine as determined by specific amplification of 16S ribosomal DNA. Applied Environ. Microbiol., 68: 114-123.
CrossRefDirect Link - Kok, R., K. Waal, A. Schut, F. Welling, G.W.G. Weenk and K.J. Hellingwerf, 1996. Specific detection and analysis of a probiotic Bifidobacterium strain in infant feces. Applied Environ. Microbiol., 62: 3668-3672.
Direct Link - Kaufmann, P., A. Pfefferkorn, M. Teuber and L. Meile, 1997. Identification and quantification of Bifidobacterium species isolated from food with genus specific 16S rRNA targeted probes by colony hybridization and PCR. Applied Environ. Microbiol., 63: 1268-1273.
Direct Link - Ke, D., C. Menard, F.J. Picard, M. Boissinot, M. Ouellette, P.H. Roy and M.G. Bergeron, 2000. Development of conventional and real-time PCR assays for the rapid detection of group B streptococci. Clin. Chem., 46: 324-331.
Direct Link - Kaplan, C.W., J.C. Astaire, M.E. Sanders, B.S. Reddy and C.L. Kitts, 2001. 16S ribosomal DNA terminal restriction fragment pattern analysis of bacterial communities in feces of rats fed Lactobacillus acidophilus NCFM. Applied Environ. Microbiol., 67: 1935-1939.
CrossRefDirect Link - Lan, P.T.N., H. Hayashi, M. Sakamoto and Y. Benno, 2002. Phylogenic analysis of cecal microbiota in chicken by the use of 16S rDNA clone libraries. Microbiol. Immunol., 46: 371-382.
Direct Link - Lee, L.G., C.R. Connel and W. Bloch, 1993. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res., 21: 3761-3766.
Direct Link - Martin, F.E., M.A. Nadkarni, N.A. Jacques and N. Hunter, 2002. Quantitative microbiological study of human carious dentine by culture and real-time PCR: Association of anaerobes with histopathological changes in chronic pulpitis. J. Clin. Microbiol., 40: 1698-1704.
CrossRefDirect Link - Nogva, H.K., A. Bergh, A. Holck and K. Rudi, 2000. Application of the nuclease PCR assay in evaluation and development of method for quantitative detection of Campylobacter jejuni. Applied Environ. Microbiol., 66: 4029-4036.
Direct Link - Reysenbach, A.L., L.J. Giver, G.S. Wickham and N.R. Pace, 1992. Differential amplification of rRNA genes by polymerase chain reaction. Applied Environ. Microbiol. 58: 3417-3418.
Direct Link - Rauter, C., R. Oehme, I. Diterich, M. Engele and T. Hartung, 2002. Distribution of clinically relevant Borrelia genospecies in ticks assessed by a novel, single-run, real-time PCR. J. Clin. Microbiol., 40: 36-43.
CrossRefDirect Link - Requena, T., J. Burton, T. Matsuki, K. Munro and M.A. Simon et al., 2002. Identification, detectionand enumeration of human Bifidobacterium species by PCR targeting the transaldolase gene. Applied Environ. Microbiol., 68: 2420-2427.
Direct Link - Salanitro, J.P., I.G. Fairchilds and Y.D. Zgornicki, 1974. Isolation, culture characteristics and identification of anaerobic bacteria from the chicken cecum. Applied Microbiol., 27: 678-687.
CrossRefDirect Link - Suzuki, M.T. and S.J. Giovannoni, 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Applied Environ. Microbiol, 62: 265-630.
Direct Link - Stiles, M.E. and W.H. Holzapfel, 1997. Lactic acid bacteria of foods and their current taxonomy. Int. J. Food Microbiol., 36: 1-29.
CrossRefPubMedDirect Link - Suzuki, M.T., L.T. Taylor and E.F. De Long, 2000. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5'-nuclease assay. Applied Environ. Microbiol., 66: 4605-4614.
Direct Link - Tilsala-Timisjarvi, A. and T. Alatossava, 1997. Development of oligonucleotide primers from the 16S-23S rRNA intergenic sequences for identifying different dairy and probiotic lactic acid bacteria by PCR. Int. J. Food Microbiol., 35: 49-56.
Direct Link - Tondella, M.L.C., D.F. Talkington, B.P. Holloway, S.F.D. Owell and K. Cowley et al., 2002. Development and evaluation of real-time PCR-based fluorescence assays for detection of Chlamydia pneumoniae. J. Clin. Microbiol., 40: 575-583.
CrossRefDirect Link - Walter, J., C. Hertel, G W., C.M. Tannock, K. Munro and W.P. Hammes, 2001. Detection of Lactobacillus, Pediococcus, Leuconostoc and Weissella species in human feces by using group-specific PCR primers and denaturing gradient gelelectrophoresis. Applied Environ. Microbiol., 67: 2578-2585.
CrossRefDirect Link - Wang, R.F., W.W. Cao and C.E. Cerniglia, 1997. PCR detection of Ruminococcus spp. in human and animal faecal samples. Mol. Mol. Cell. Prob., 11: 259-265.
Direct Link - Wang, R., W. Cao and C.E. Cerniglia, 1996. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Applied Environ. Microbiol., 62: 1242-1247.
Direct Link - Wilson, K.H. and R.B. Blitchington, 1996. Human colonic biota studied by ribosomal DNA sequence analysis. Applied Environ. Microbiol., 62: 2273-2278.
Direct Link - Yamamoto, T., M. Morotomi and R. Tanaka, 1992. Species-specific oligonucleotide probe for five Bifidobacterium species detected in human intestinal microflora. Applied Environ. Microbiol., 58: 4076-4079.
Direct Link - Zoetendal, E.G., A.D.L. Akkermans and W.M. De Vos, 1998. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Applied Environ. Microbiol., 64: 3854-3859.
PubMed