
Nihal Buzkan
Not Available
Andrew Walker
Not Available
Asian Journal of Plant Sciences
Year: 2004 | Volume: 3 | Issue: 3 | Page No.: 387-390







ABSTRACT
Detection of Grapevine fanleaf virus (GFLV) in grapevine samples is difficult due to seasonal effect, low viral titer and uneven distribution of the viral particles within the plant and presence of phenolic compounds. To circumvent this problem in grape tissue, a nucleic acid purification (viral RNA) procedure is aimed to improve the extraction methods to remove the inhibitory components of the tissue. In this work, RNA from hybrid grapevines of five different populations (9621, 9623, 9630, 9631 and 9635) from the breeding program of the Department of Viticulture and Enology, University of California, Davis (CA) on which nematodes (Xiphinema index) carrying GFLV was extracted with silica particle suspension and phenol/chloroform methods. Extracted RNAs were submitted to cDNA synthesis and amplification procedure with primers specific to GFLV. The silica method could detect the virus infection from 43 cuttings although viral nucleic acid of the same samples from phenol/chloroform extraction were not amplified in PCR reaction The success of the silica methods was because phenolic compounds were bound to silica particles, eliminated by buffer solution with ethanol. The protocol does not require ultracentrifugation and is possible to complete in few hours which is much shorter than phenol/chloroform application. It also appears to be widely applicable to particularly difficult plant tissues.
PDF Abstract XML References Citation
How to cite this article
Nihal Buzkan and Andrew Walker, 2004. A Small-scale Procedure for Extracting Nucleic Acids from Grapevine Dormant Cuttings Infected with GFLV. Asian Journal of Plant Sciences, 3: 387-390.
DOI: 10.3923/ajps.2004.387.390
URL: https://scialert.net/abstract/?doi=ajps.2004.387.390
DOI: 10.3923/ajps.2004.387.390
URL: https://scialert.net/abstract/?doi=ajps.2004.387.390
INTRODUCTION
Fanleaf degeneration is an economically important disease that confronts grape growers worldwide. It is caused by grapevine fanleaf virus (GFLV), a member of the nepovirus group, transmitted in the soil by the longidorid nematodes, Xiphinema index Thorne and Allen (dagger nematode)[1]. This disease greatly reduces crops due to poorly filled clusters with numerous unfertilized “shot” berries, also induces leaf deformation, yellow mosaic and vein banding[2]. In some vineyards soil disinfection using nematicides remained inefficient. Furthermore use of nematicides is being restricted because of the detrimental effects on the soil inhabiting fauna and on dripping water. Therefore new approaches for introducing virus resistance to grapevine would be desirable[3,4]. Efforts to produce fanleaf-resistant rootstocks commenced at the University of California, Davis, with an assessment of Vitis species for resistance to X. index feeding[5]. Moreover, rapid and reliable detection of the virus and its causal agent is essential. The development of a reverse transcriptase-polymerase chain reaction (RT-PCR) assay[6] provided a virus detection system with the sensitivity to potentially overcome some of the afore-mentioned difficulties. But the general application of this approach to the detection of GFLV in woody grapevine samples is often blocked due to seasonal effect, low viral titer and uneven distribution of the viral particles within the plant and the colored oxidizing, cross-linking nature of woody plant extracts because of phenolic compounds[7,8]. To circumvent this problem in grape tissue, a nucleic acid purification (viral RNA) procedure can be developed to remove the inhibitory components of the tissue extract.
In this work we applied two different nucleic acid extraction methods to woody cuttings of hybrid grapevine on which viruliferous nematodes were fed and compared the results to improve reliable and sensitive way for GFLV RNA detection.
MATERIALS AND METHODS
Grapevine and viral source: Interspecific hybrids (Vitis rupestris x Muscadinia rotundifolia) from breeding programs of Prof. A. Walker at the Department of Viticulture and Enology, University of California, Davis (CA) on which viruliferous nematodes were fed (100 nematodes/plant) were tested for nematode resistance. V. rupestris cv. St. George and M. rotundifolia cv. Trayshed were susceptible and resistant control, subsequently. Dormant cuttings from each hybrid were collected from October through December 2000, labelled carefully and stored at +4°C until their use.
RNA extraction: Viral particles were extracted from phloem scrapings with phenol/chlorofm[9] and silica particle suspension methods.
For phenol/chloroform extraction, about 100 mg of scraped bark tissue from dormant cuttings was powdered in a mortar and pestle with the presence of liquid nitrogen until the tissue became light green. Powdered tissue was extracted with 1.5 vol. of 1X GES buffer solution (0.1M glycine-NaOH pH 9.0, 50 mM NaCl, 1mM EDTA), mixed well with equal volume of Tris-saturated phenol/chloroform (1:1). The aqueous phase was mixed thoroughly with chloroform. Clarified extracts were saturated with 2.5 vol. of cold 95% ethanol and 0.1 vol. of 3 M sodium acetate (pH 5.5) either at -20°C for 3 h or -80°C overnight. Subsequent centrifugation was carried out to collect the pellet at 13,000 rpm. for 10-15 min., washed with 70% cold ethanol. The pellet was lyophilized and resuspended in TE buffer (10 mM Tris-HCl, 1 mM EDTA pH 7.5), stored at -70°C for PCR use.
For silica method, phloem scrapings (100 mg) were ground with grinding buffer (4M guanidine thiocyanate, 2 M sodium acetate pH 5.2, 25 mM EDTA, 1 M potassium acetate, 2.5% PVP-40) and 1% of β-mercaptoethanol. Aqueous part of 500 μl was recovered and incubated at 70°C for 10 min. with 10% sodium sarcosyl, subsequently transferred into ice for 5 min. The mixture was centrifuged at 13,000 rpm for 10 min., then saturated with 150 μl of 95% ethanol, 300 μl 6M NaI, 25 μl of resuspended silica at room temperature. Collected pellets were washed twice with washing buffer (10 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 50 mM NaCl, 50% ethanol), resuspended in sterile distilled water.
Extracted RNAs were measured with spectrophotometer reading at 260/280 nm.
Reverse transcription and complementary DNA (cDNA) synthesis: cDNA synthesis was performed essentially as described by Hadidi and Yang[10]. Briefly, the sample volume was brought to 30 μl with 1X first strand cDNA buffer (GIBCO-BRL) after the sample was boiled for 5 min. at 95°C. After a 30 min annealing at room temperature, 19 μl of reaction solution (containing 4 μl 5X first strand buffer, 5 μl 0.3 M mercaptoethanol, 2.5 μl 10 mM dNTP, 1 μl RNAsin (40 U μl-1, Promega) and 6.5 μl water) and 1 μl cloned M-MuLV-transcriptase (200 U μl-1, GIBCO BRL) were added and the mixture was incubated for 1 h at 37°C.
Polymerase chain reaction: The primers for GFLV were of the coat protein region located at the 3’ end of RNA2 between nucleotides 762 and 1,083 with the sequences: 5’ CCAAAGTTGGTTTCCCAAGA 3’ (antisense primer) and 5’ ACCGGATTGACGTGGGTGAT 3’ (sense primer)[11]. Five μl aliquot of cDNA reaction was added into 45 μl of the amplification mixture (vol/reaction: 5 μl 10X thermophilic buffer, 2 μl 25 mM MgCl2, 1.5 μl 10 mM dNTP, 1.5 μl 120 nM sense and antisense primers) with 1U of DNA Taq polymerase (GIBCO BRL) and placed in thermo cycler (Perkin-Elmer, Cetus). PCR amplification was performed for 1 min at 95°C (denaturation), 1 min at 53°C (annealing), for 1 min at 72°C (primer extension for 35 cycles and final extension at 72°C for 7 min.
Analysis of PCR amplified products: Aliquots (5 μl) of the PCR amplified cDNA fragments were analyzed by electrophoresis through 1.2% agarose gel at 5V cm-1 for 90 min in 1X TBE buffer (89 mM Tris, 89 mM boric acid, 2.5 mM EDTA pH 8.0). Separated DNA fragments were visualized with ethidium bromide staining (10 mg ml-1) and photographed.
RESULTS
Total of 139 plants of five different populations (9621, 9623, 9630, 9631 and 9635) from the breeding program of the Department of Viticulture and Enology, University of California, Davis (CA) was used for RNA extraction. When viral RNAs from phenol/chloroform method were checked for quality purpose in 1.2% agarose gel electrophoresis, a strong background caused by plant proteins, carbonhydrates and phenolic compounds in the gel. RNAs were exposed to further cleaning with chloroform application. It was still very diffult to eliminate all the contaminants and the quantity of extracted RNA strongly dropped after second treatment. After the first PCR application, no signs for cDNA amplification regarding GFLV coat protein gene were obtained (not shown).
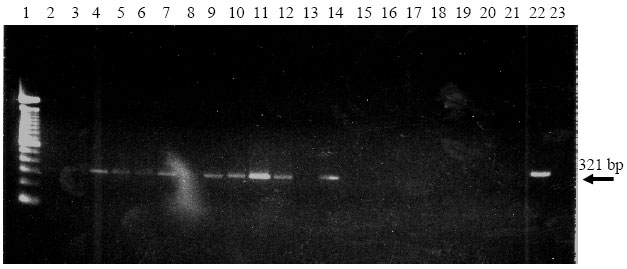 | |
| Fig. 1: | Electrophoretic analysis of the amplified cDNA (321 bp) of the extracted RNAs from hybrid population with silica particle suspension (2-14) and phenol/chloroform (15-21) methods. Lane 1: 100 bp DNA standard, 22: RNA from naturally GFLV-infected grapevine extracted with silica particle suspension method; 23: RNA from naturally GFLV-infected grapevine extracted with phenol/chloroform method |
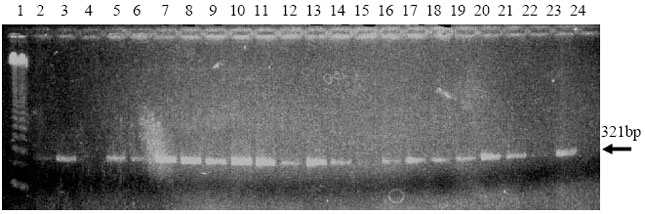 | |
| Fig. 2: | Electrophoretic analysis of the amplified cDNA of RNA extracted with silica particle suspension method from GFLV infected hybrid populations. The size of the DNA bands was 321 bp. Lane 1: 123 bp DNA standard; Lane 2-7: 9621; Lane 8-13: 9623; Lane 14-19: 9631; Lane 20-22: 9635; Lane 23: RNA from naturally GFLV-infected grapevine 24: water control. |
Detailed trials were set up to increase the amplification possibility such as; (I) increase of initial RNA amount for reverse transcription and cDNA synthesis and/or; (ii) increase of cDNA amount in the PCR mixture. In both cases, the amplification of cDNA from infected grapevine plants including positive controls was unsuccessful (Fig. 1). This would be due to the lack of good quality and quantity of the extracted RNAs with phenol/chloroform treatment to perform reverse transcription step.
The RNAs obtained with silica particle suspension method coming from the same plants were used for the amplification process in the same amount as described for those from phenol/chloroform treatment. However, none of the samples was amplified, neither did positive control. Once the amount of RNA was increased to 10 μl for reverse transcription, clear DNA bands at 321 bp in size were observed after electrophoretic analysis (Fig. 1). Quantity of viral RNA from silica particle suspension method was found to be higher than those from phenol/chloroform method. All GFLV infected grapevine plants were routinely tested and 43 out of 139 plants were found to be carrying GFLV. Infection ratio varied within the populations; however, the highest ratio was in 9623 (Fig. 2).
DISCUSSION
Specificity, sensitivity and versatility are three reasons that PCR has generated considerable interest as a diagnostic tool for GFLV detection infected grapevine. However, PCR has its limitations because the viral titer in infected grapevine tissue is low and grapevine is rich in phenolic compounds which interfere with assays. Because very small amounts of nucleic acid are needed for GFLV amplification by PCR, the development of a rapid, small-scale procedure would allow testing many samples and increase the efficacy of the method.
It was possible to extract viral RNA in good quality and quantity with silica particle suspension method that was particularly convenient for extracting small amounts of tissues. Using this method, large number of field samples can be collected and processed immediately or stored at cold for later testing. Amplification results demonstrated successful detection of GFLV from the extracted RNA; however, PCR did not work with the RNAs coming from phenol/chlorofm application. Plant phenolic compounds are bound to silica particles and removed from the RNA suspension. This method is found to be quite simple and can be concluded within 2-3 h respect to the other one with long precipitation step at -20°C for 3 h or -80°C overnight. No specific experience is necessary for handling the silica particle suspension method. Finally, the chemicals and stock reagents were relatively inexpensive compared to commercially available nucleic acid extraction kits.
On the other hand, use of phenol/chloroform needs technical experience and great care during the protocol application due to their toxic nature to human health. Whole procedure takes more time as compared to silica particle suspension method.
ACKNOWLEDGMENT
Special thanks to Prof. Andrew Walker for his review and critical comments on the manuscript.
REFERENCES
- Raski, D.J., A.C. Goheen, L.A. Lider and C.P. Meredith, 1983. Strategies against grapevine fanleaf virus and its vector nematode. Plant Dis., 67: 335-339.
Direct Link - Raski, D.J. and A.C. Goheen, 1988. Comparison of 1,3-dichloropropene and methylbromide for control of Xiphinema index and grapevine fanleaf degeneration complex. Am. J. Enol. Viticul., 39: 334-336.
Direct Link - Walker, M.A., E. Weber and J.A. Wolpert, 1994. Fiel screening of grape rootstock selections for resistance to fanleaf degeneration. Plant Dis., 78: 134-136.
Direct Link - Rowhani, A., M.A. Wlaker and S.S. Rokni, 1992. Sampling strategies for the detection of grapevine fanleaf virus and the grapevine strain of tomato ringspot virus. Vitis, 31: 35-44.
Direct Link - Demeke, T. and R.P. Adams, 1992. The effects of plant polysaccharide and buffer additives on PCR. Biotechniques, 12: 332-334.
PubMed - White, J.L. and J.M. Kaper, 1989. A simple method for detection of viral satellite RNAs in small plant tissue samples. J. Virol. Method, 23: 83-93.
Direct Link - Hadidi, A. and X. Yang, 1990. Detection of pome fruit viroids by enzymatic cDNA amplification. J. Virol. Methods, 30: 261-269.
Direct Link - Sanchez, F., C. Chay, M.J. Borja, A. Rowhani, J. Romero, G. Bruening and F. Ponz, 1991. cDNA sequences of the capsid protein gene and 3! un translated region of a fanleaf isolate of grapevine fanleaf virus. Nucl. Acids Res., 19: 5440-5440.
Direct Link