
A.R. Tapas
Department of Pharmaceutical Chemistry, Sudhakarrao Naik Institute of Pharmacy, Pusad-445204, Dist: Yavatmal, Maharashtra, India
LiveDNA: 91.6535
P.S. Kawtikwar
Shri Sureshdada Jain Institute of Pharmaceutical Education and Research, Jamner-424206, Dist: Jalgaon, Maharashtra, India
D.M. Sakarkar
Department of Pharmaceutical Chemistry, Sudhakarrao Naik Institute of Pharmacy, Pusad-445204, Dist: Yavatmal, Maharashtra, India
American Journal of Drug Discovery and Development
Year: 2011 | Volume: 1 | Issue: 3 | Page No.: 160-173







ABSTRACT
The objective of the present work was to enhance the solubility, dissolution rate and micromeritic properties of Felodipine (FL) a poorly water soluble antihypertensive, using spherical agglomeration by Quasi Emulsion Solvent Diffusion (QESD) method. In this study methanol, water and dichloromethane were used as good solvent, poor solvent and bridging liquid, respectively. The hydrophilic polymers like Hydroxypropyl β-cyclodextrin, β-cyclodextrin, Lutrol F68, Lutrol F127 were used in agglomeration process. The pure drug (FL) and its agglomerates with different polymers were characterize for their drug loading as well as by Differential Scanning Calorimetry (DSC), X-ray Diffraction (XRD), FTIR spectroscopic studies and Scanning Electron Microscopy (SEM). The DSC results indicated decrease in melting enthalpy related to disorder in the crystalline content. XRD studies also showed changes in crystallanity, IR spectroscopy revealed that there were no chemical changes in the recrystallized agglomerates. The spherically agglomerated solid dispersions with different polymers exhibited marked increase in solubility (53.69±0.53 μg mL-1), dissolution rate (100.09±2.27% in 60 min) and micromeritic properties (bulk density, flow property, compactability) compared with FL. The SEM studies showed that the agglomerate possesses a good spherical shape.
PDF Abstract XML References Citation
Received: February 19, 2011;
Accepted: May 26, 2011;
Published: June 18, 2011
How to cite this article
A.R. Tapas, P.S. Kawtikwar and D.M. Sakarkar, 2011. Modification of Felodipine Properties using Spherically Agglomerated Solid Dispersions. American Journal of Drug Discovery and Development, 1: 160-173.
DOI: 10.3923/ajdd.2011.160.173
URL: https://scialert.net/abstract/?doi=ajdd.2011.160.173
DOI: 10.3923/ajdd.2011.160.173
URL: https://scialert.net/abstract/?doi=ajdd.2011.160.173
INTRODUCTION
Research and development productivity upgrading is one of the biggest tasks facing the pharmaceutical industry. Advances in genomics and proteomics in recent years have led to a flare-up in the number of possible drug targets and so pharmaceutical companies have made major investments in high throughput screening and combinatorial chemistry to identify more potential drug candidates for these novel targets. Many of the drugs, evolving from these techniques, can be categorized as class II according to the Biopharmaceutical Classification System (BCS) (Lobenberg and Amidon, 2000). These drugs are poorly water soluble but once they are dissolved they are easily absorbed through the gastro-intestinal membrane. One of the approaches to enhance the dissolution rate is the use of solid dispersions. At present the solvent method and melting method are widely used in the preparation of solid dispersions (Habib et al., 2000; Vasconcelos et al., 2007; Sharma and Joshi, 2007). In general, subsequent grinding, sieving, mixing and granulation are necessary to produce the different desired formulations. The spherical agglomeration technique has been used as an efficient particle preparation technique (Gupta et al., 2007; Usha et al., 2008; Yadav and Yadav, 2008).
 |
| Fig. 1: | Chemical structure of felodipine |
Initially, spherical agglomeration technique was used to improve powder flowability and compressibility (Kawashima et al., 1994; Bodmeier and Paeratakul, 1989). Then polymers were introduced in this system to modify their release (Di-Martino et al., 1999). Currently, this technique is used more frequently for the solid dispersion preparation of water-insoluble drugs in order to improve their solubility, dissolution rate and simplify the manufacturing process (Cui et al., 2003). Spherical crystallization (Kawashima et al., 1982) has been developed as a novel particulate design technique to improve processability such as mixing, filling, tableting characteristics and dissolution rate of pharmaceuticals. The resultant crystals can be designated as spherical agglomerates (Kulkarni and Nagavi, 2002). Spherical crystallization is an effective alternative to improve dissolution rate of drugs (Sano et al., 1992). This can be achieved by various methods such as spherical agglomeration, Quasi-Emulsion Solvent Diffusion (QESD) and neutralization methods out of which QESD is most commonly used (Kawashima, 1984). When bridging liquid plus good solvent containing drug are poured into the poor solvent under agitation, quasi-emulsion droplets of bridging liquid or good solvent form in the poor solvent and induces crystallization of the drug followed by agglomeration (Yang et al., 2003; Tapas et al., 2009, 2010).
Felodipine (FL) (Fig. 1), a second-generation calcium antagonist of the 1,4-Dihydropyridine (DHP) type, lowers blood pressure by selective dilation of arterial smooth muscles in peripheral resistance vessels (Saltiel et al., 1988). Clinical studies have demonstrated that felodipine which is approved for marketing in several countries, is an effective, well tolerated antihypertensive drug (Dunselman and Edgar, 1991). The major problem of felodipine is its very low water solubility which results into poor dissolution rate (Karavas et al., 2001).
In the present study, to overcome the problems related to solubility, dissolution rate, flowability and compressibility, the spherically agglomerated solid dispersions of FL were prepared by QEDS which is more convenient and is cheaper. In addition, incorporating hydrophilic polymers (Hydroxypropyl β-cyclodextrin, β-cyclodextrin, Lutrol F68, Lutrol F127) during agglomeration imparted better solubility, dissolution rate, flowability and compressibility.
MATERIALS AND METHODS
Materials: Felodipine USP was generously provided as a gift sample from Cipla Ltd., Mumbai Central, Mumbai, India. Lutrol F68 and Lutrol F127 were obtained as a gift sample from BASF Pharma, Mumbai, India. Hydroxy propyl β-cyclodextrin (HPβCD) and β-cyclodextrin (βCD) were obtained as a gift sample from Signet Chemical Corporation Pvt. Ltd., Mumbai, India.
| Table 1: | Composition of spherical agglomerates |
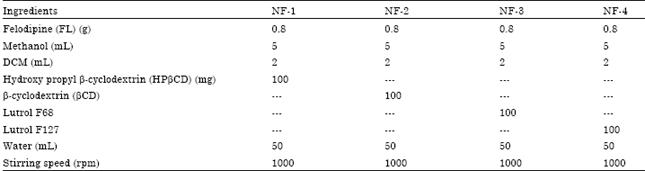 | |
Methanol and dichloromethane were purchased from Lobachemie, Mumbai, India. All other chemicals used were of analytical grade.
Methods: All spherical agglomerated solid dispersions were obtained by the quasi emulsion solvent diffusion method. Felodipine (0.8 g) with various polymers was dissolved in good solvent methanol (5.0 mL). The bridging liquid dichloromethane (2.0 mL) was added to it. The resulting solution was then poured dropwise in to the poor solvent distilled water (50 mL). The mixture was stirred continuously for a period of 0.5 h using a controlled speed mechanical stirrer (Remi motors, India) at 1000 rpm to obtain spherically agglomerated solid dispersion. As the good solvent diffused into the poor solvent, droplets gradually solidified and formed spherically agglomerated solid dispersion. The agglomerates were separated by filtration using Whatman filter paper (No.1) and dried in desiccator at room temperature. The amount of polymers was altered to get desired agglomerates. The composition is given in the Table 1.
Infrared spectroscopy, differential scanning calorimetry (DSC) and Powder X-ray diffraction studies (PXRD): The Infrared (IR) spectra of powder Felodipine and the agglomerates were recorded on an IR-spectrophotometer (IRAFFINITY-1, Shimadzu, Japan). Differential Scanning Calorimetry (DSC) analysis was performed using a DSC 823 calorimeter (Mettler Toledo model) operated by star e software. Samples of Felodipine and its agglomerates were sealed in an aluminium crucible and heated at the rate of 10°C min-1 up to 300°C under a nitrogen atmosphere (40 mL min-1). Powder X-ray diffraction patterns (XRD) of the felodipine and spherical agglomerates were monitored with an X-ray diffractometer (Panalytical Xpert pro MPD xrd machine) using copper as X-ray target, a voltage of 40 kV, a current of 30 mA and with 1.5404 Angstorm wavelength. Xcelerator RTMS with secondary monochromator was used as a detector. The samples were analyzed over 2θ range of 7.02-59.98° with scanning step size of 0.02° (2θ) and scan step time of one second.
Micromeritic properties: The size of agglomerates was determined by microscopic method using stage and eyepiece micrometers. The shape of the agglomerates was observed under an optical microscope (60x magnification) attached to a computer. The Loose Bulk Density (LBD) and Tapped Bulk Density (TBD) of plain felodipine and its spherical agglomerates were determined. Carr’s index and Hausner’s ratio were calculated using LBD and TBD values (Wells, 2002). The angle of repose was accessed by the fixed funnel method (Martin et al., 2002).
Scanning electron microscopy: The surface morphology of the agglomerates was accessed by Scanning Electron Microscopy (SEM). The crystals were splutter coated with gold before scanning.
Drug loading: The drug loading efficiency of agglomerates was determined by dissolving 100 mg of crystals in 5 mL methanol and diluting further with distilled water (100 mL), followed by measuring the absorbance of appropriately diluted solution spectrophotometrically (PharmaSpec UV-1700, UV-Vis spectrophotometer, Shimadzu) at 362 nm.
Solubility studies: A quantity of crystals (about 100 mg) was shaken with 10 mL distilled water in an incubator shaker for 24 h at room temperature. The solution was then passed through a whatmann filter paper (No. 42) and amount of drug dissolved was analyzed spectrophotometrically.
In vitro dissolution studies: The in vitro dissolution studies were carried out using an 8 station USP 23 dissolution testing apparatus (Electrolab, India). The dissolution medium used was 900 mL of Phosphate buffer pH 6.8 containing 1.8% Tween 80 (Yan et al., 2009). The dissolution medium was kept at in a thermostatically controlled water bath at 37±0.5°C. The agglomerates and pure drug containing 10 mg of Felodipine were weighed and introduced into the dissolution medium. The medium was stirred at 75 rpm using paddle. The dissolution tests were carried out for 60 min. At predetermined time intervals 5 mL of samples were withdrawn and analyzed spectrophotometrically. At each time of withdrawal, 5 mL of fresh corresponding medium was replaced into the dissolution flask. The cumulative amount of drug release was calculated and plotted versus time.
Dissolution efficiency studies: The dissolution efficiency of the batches was calculated by the method mentioned by Khan (1975). It is defined as the area under the dissolution curve between time points t1 and t2 expressed as a percentage of the curve at maximum dissolution, y100, over the same time period or the area under the dissolution curve up to a certain time, t (measured using trapezoidal rule) expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time (Anderson et al., 1998).
DE60 values were calculated from dissolution data and used to evaluate the dissolution rate.
Accelerated stability study: The agglomerates were charged for the accelerated stability studies in accordance with ICH guidelines (40±2°C and 75±5% RH for a period of six months). The initial drug content of optimized agglomerates was determined. Then they were placed in USP type I flint glass vials, closed with bromobutyl rubber bung and sealed with aluminium caps. The vials were placed in stability chamber (Thermolab, Mumbai, India) and samples were withdrawn at predetermined time intervals (15, 30, 60, 90 and 180 days) and evaluated for drug content.
Statistical analysis: The results were analyzed by two tailed Student’s t-test using Graph Pad Instat software (GPIS; version 5.0) and Microsoft Excel 2007. The results obtained from the dissolution studies were statistically validated using ANOVA (Dunnett Multiple Comparisons Test). The Dissolution Efficiency (DE) was calculated using the origin software (Origin pro; version: 8.1).
RESULTS AND DISCUSSION
Formulation and development: Spherically agglomerated solid dispersion of Felodipine were prepared by Quasi Emulsion Solvent Diffusion method (QESD) using a three solvent system. It involves good solvent, poor solvent and a bridging liquid. The selection of these solvents depends on the miscibility of the solvents and the solubility of drug in individual solvent. Accordingly methanol, dichloromethane, water were selected as a good solvent, bridging liquid and poor solvent, respectively. Felodipine is soluble in methanol but poorly soluble in water. Also it is soluble in dichloromethane which is immiscible in water. Hence, this solvent system was used in the present study. In QESD method, when good solvent solution of drug plus bridging liquid were poured in the poor solvent under agitation, quasi emulsion droplets of bridging liquid and good solvent were produced. Then the good solvent diffuses gradually out of the emulsion droplet into the outer poor solvent phase. The counter-diffusion of the poor solvent into the droplets induces the crystallization of the drug within the droplet due to the decrease in solubility of the drug in the droplet containing the poor solvent. The bridging liquid wets the crystal surface to cause binding and promotes the formation of liquid bridges between the drug crystals to form spherical agglomerates. The spherically agglomerated crystals are formed by coalescence of these dispersed crystals.
FTIR, DSC and PXRD studies: The possible interaction between the drug and the carrier was studied by IR spectroscopy. The infrared spectra of felodipine as well as its spherically agglomerated solid dispersions are presented in Fig. 2(a-e). IR spectra of felodipine showed characteristic peaks at 3371.57 (N-H Str., Secondary), 2989.66 (C-H Str., -CH3), 3068.75 (C-H Str., Aromatic), 1689.64 (C = O Str.), 769.60 (C-Cl Str.) cm-1. There were no considerable changes in the IR peaks of the spherical agglomerates when compared to pure felodipine. If there is any strong interaction between drug and carrier, it often leads to identifiable changes in the IR profile and melting point of the drug. The results of IR spectra indicated the absence any well-defined interaction between felodipine and HPβCD, βCD, Lutrol F68, Lutrol F127 in presence of methanol, dichloromethane and water.
The DSC thermograms of pure Felodipine and its spherical agglomerates are presented in Fig. 3(a-e). The DSC thermogram of Felodipine showed a single endotherm at 146.28°C which was ascribed to drug melting. There was a negligible change in the melting endotherms of prepared spherical agglomerates compared to pure drug (NF1 = 146.14°C, NF2 = 146.12°C, NF3 = 146.18°C, NF4 = 144.62°C). In NF3 and NF4 the endotherm at 56.94°C and 56.82°C was ascribed to the melting of Lutrol F68 and Lutrol F127, respectively. This observation further supports the IR spectroscopy results which indicated the absence of any interactions between the drug and additives used in the preparation. However, there was a decrease, although very small, in the melting point of the drug in the spherical agglomerates compared to that of pure felodipine. This indicates the little amorphization of felodipine when prepared in the form of agglomerates.
The results of powder X-ray diffraction pattern of felodipine and spherical agglomerates are shown in Fig. 4(a-e). Pure drug exhibited intense and long peaks whereas spherical agglomerates showed a halo pattern with less intense peaks which indicate a considerable decrease in crystallinity or partial amorphization of the drug in its agglomerated form. The percentage of crystallinity will be directly related to the ratio between the integrated intensities caused by the crystalline and amorphous material in a similar measurement range of XRPD diffractogram (Bates and Ivanisevic, 2005). However, a standard method for measuring the crystalline percentage of a mixture is using the standard proportion of an amorphous powder in combination with the studied drug.
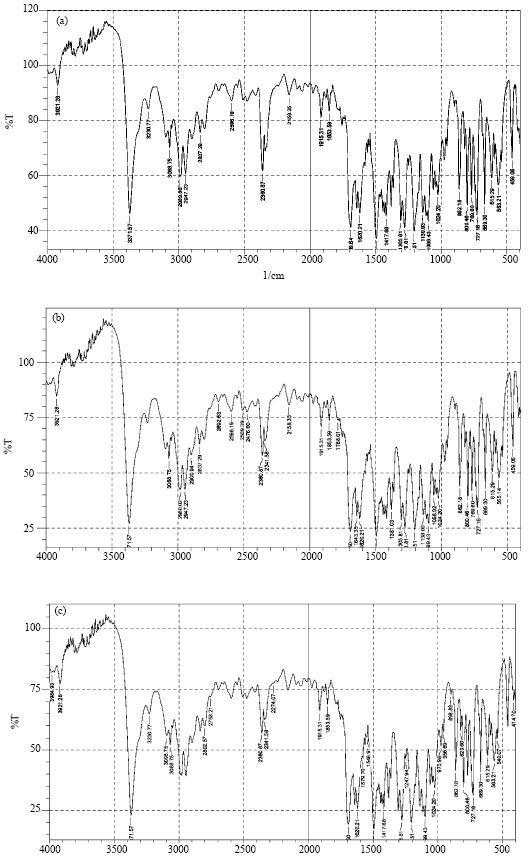 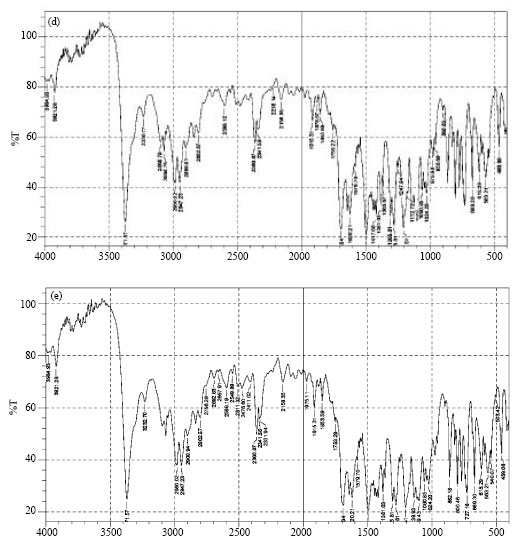 |
| Fig. 2(a-e): | IR spectra of (a) felodipine (b) spherical agglomerates NF1, (c) spherical agglomerates NF2, (d) spherical agglomerates NF3 and (e) spherical agglomerates NF4 |
By plotting standard curves, the amount of unknown crystalline percentage may be calculated. These results further support the DSC data which demonstrated partial amorphization of the drug agglomerates.
Micromeritics properties: The mean particle diameter of agglomerates is shown in Table 2. The pure drug exhibited a very small particle size (83.12±10.21 μm, n = 3) whereas the size of prepared agglomerates was found between 148.00±10.58 and 712.00±14.52 μm, n = 3 which is significantly different from that of pure drug (p<0.05). The shape of the crystals (not shown), when observed using an optical microscope was spherical in all the prepared agglomerated formulation.
The results of Loose Bulk Density (LBD), Tapped Bulk Density (TBD), Carr’s index, Hausner’s ratio, angle of repose are presented in Table 2. These parameters were used to assess the packability, flow and compressibility properties of the agglomerates. The LBD, TBD, Carr’s index, Hausner’s ratio and angle of repose values for pure drug FL were 0.39±0.02 g mL-1( n = 3), 0.62±0.01 g mL-1 (n = 3), 37.60±0.50% (n = 3), 1.60±0.03 (n = 3), 51.34±0.32° (n = 3), respectively, indicating poor flow and packability properties.
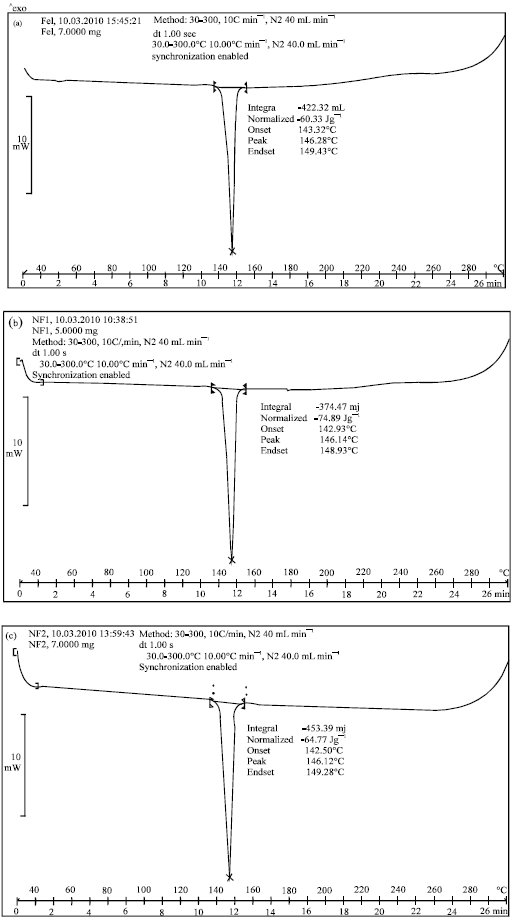 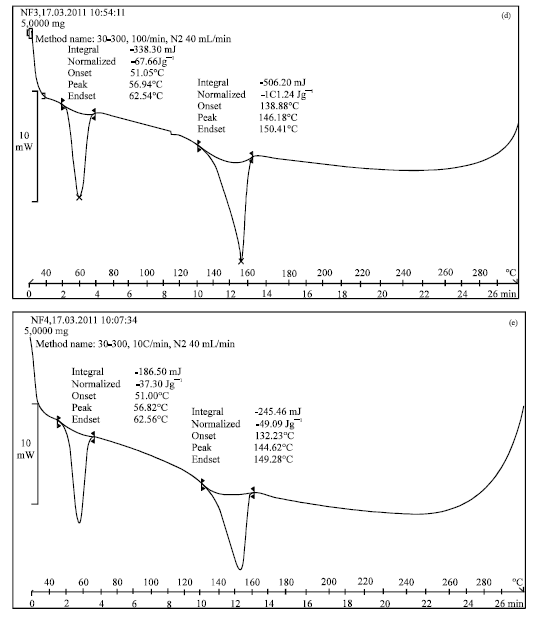 |
| Fig. 3(a-e): | DSC Patterns of (a) felodipine (b) spherical agglomerates NF1, (c) spherical agglomerates NF2, (d) spherical agglomerates NF3, (e) spherical agglomerates NF4 |
On the other hand, all prepared spherical agglomerates exhibited higher LBD (0.43±0.01 to 0.47±0.01 g mL-1, n = 3) and TBD (0.48±0.01 to 0.57±0.02 g mL-1, n = 3) values which indicate good packability. Also all the prepared agglomerates exhibited low Carr’s index, Hausner’s ratio and angle of repose values, indicating excellent flow properties and compressibility (Carr’s index: 08.21±5.41 to 16.60±4.33%, n = 3; Hausner’s ratio: 1.09±0.06 to 1.20±0.06, n = 3; angle of repose: 11.66±0.35° to 18.13±0.21°, n = 3). The improved flowability and compressibility of spherical agglomerates may be due to the sphericity, regular and larger size of crystals.
Scanning electron microscopy: The results of surface morphology studies are shown in Fig. 5 (a-e). The SEM results revealed the spherical structure of agglomerates. The surface morphology studies also revealed that the agglomerates were formed by very small crystals which were closely compacted into spherical form. These photo-micrographs show that the prepared agglomerates were spherical in shape which enabled them to flow very easily.
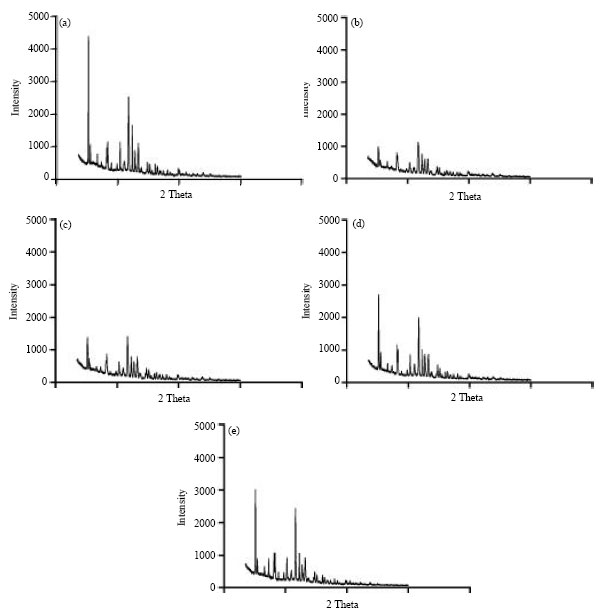 |
| Fig. 4: | X-ray diffraction spectra (a) felodipine (b) spherical agglomerates NF1, (c) spherical agglomerates NF2, (d) spherical agglomerates NF3 and (e) spherical agglomerates NF4 |
| Table 2: | Micromeritics, solubility and drug loading efficiency data for the agglomerates and pure druga |
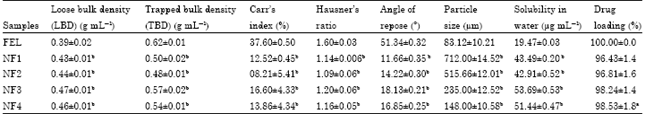 | |
| aValues are Mean±SD (n = 3). bSignificantly different compared to pure felodipine (p<0.05) | |
Drug loading and solubility studies: The results of drug loading efficiency and aqueous solubility are shown in Table 2. The drug loading of agglomerates was uniform among the different spherical agglomerates prepared and range from 96.43±1.4 to 98.53±1.8 % (n = 3), indicating negligible loss of drug during agglomeration process. The result of solubility studies indicate that pure FL possesses a very low solubility in water (19.47±0.03 μg mL-1, n = 3); the drug solubility from crystals increased significantly (p<0.05), demonstrating that the incorporation of hydrophilic polymers enhances the drug solubility.
 |
| Fig. 5(a-e): | Scanning electron micrographs of: (a) Felodipine crystals at x500, (b) spherical agglomerates containing HPβCD (NF1) at x330, (c) spherical agglomerates containing βCD (NF2) at x500, (d) spherical agglomerates containing Lutrol F68 (NF3) at x330 and (e) spherical agglomerates containing Lutrol F127 (NF4) at x55 |
Amongst the hydrophilic polymers used Lutrol F68 spherical agglomerates (NF3) shows maximum solubility (53.69±0.53 μg mL-1, n = 3).
In vitro dissolution studies: The results of in vitro dissolution studies are shown in Fig. 6 and Table 3. Pure Felodipine exhibited less release at the end of 60 min (12.20±1.84%, n = 3) while spherical agglomerates with hydrophilic polymers improved the dissolution rate of Felodipine. The agglomerates (NF3) released 100.09±2.27% (n = 3) drug at the end of 60 min. The dissolution efficiency at 30 min (DE30) for pure drug was 4.50±0.78% (n = 3), whereas for agglomerates (NF3) was 46.88±0.36% (n = 3). The results revealed that the spherical agglomerates with various polymers showed significant increase (p<0.05) in drug release compared to the pure drug. Among the different hydrophilic polymer tested, Lutrol F68 showed better effect on solubility and dissolution rate compared to other polymers. The increase in the dissolution rate of agglomerates could be attributed to deposition of polymer onto the recrystallized drug surface and better wettability of the spherical agglomerates.
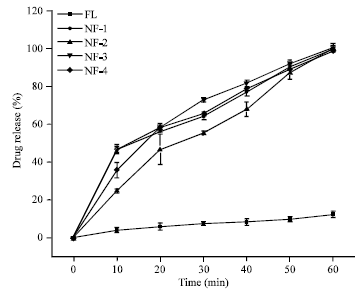 |
| Fig. 6: | Dissolution profile of pure drug and agglomerates in phosphate buffer 6.8 with 1.8% Tween 80. Values are Mean±SD (n = 3) |
| Table 3: | Drug release and dissolution efficiency |
 | |
DP60: Percent drug release at 60 min. DE30: Dissolution efficiency at 30 min. a Mean±SD, n = 3. bSignificantly different compared to pure felodipine (p<0.05) | |
| Table 4: | Results of accelerated stability study |
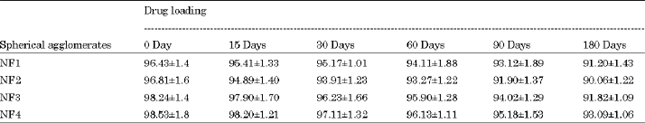 | |
Accelerated stability study: The values of drug content for spherical agglomerates were shown in Table 4. On storage the agglomerates did not show considerable change in drug content for the period of six months at accelerated condition when compared with initial value which indicates good stability of agglomerates.
CONCLUSION
The present study shows that spherically agglomerated solid dispersion of FL prepared with HPβCD, βCD, Lutrol F68 and Lutrol F127 exhibited improved solubility and dissolution rate in addition to improving the micromeritics properties. This technique may be applicable for producing oral solid dosage forms of FL with improved dissolution rate with improving physicochemical and micromeritic properties.
ACKNOWLEDGMENTS
Authors gratefully acknowledge Cipla Ltd, Mumbai Central, Mumbai, India for providing gift sample of Felodipine. Authors would like to thank AISSMS college of Pharmacy, Pune, India and Tata Institute of Fundamental Research (TIFR), Mumbai, India for their kind help, respectively, in DSC studies and PXRD studies. Also authors are thankful to Visvesvaraya National Institute of Technology (VNIT), Nagpur, India and Government college of Pharmacy, Amravati, India for providing the facilities to carryout SEM and IR analysis, respectively.
REFERENCES
- Anderson, N.H., M. Bauer, N. Boussac, R. Khan-Malek, P. Munden and M. Sardaro, 1998. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J. Pharm. Biomed. Anal., 17: 811-822.
CrossRef - Bates, S. and I. Ivanisevic, 2005. Percent crystallinity determination from XRPD data using standardless methods. The AAPS J., 7: 1-1.
Direct Link - Bodmeier, R. and O. Paeratakul, 1989. Spherical agglomerates of water-insoluble drugs. J. Pharm. Sci., 78: 964-967.
PubMed - Cui, F., M. Yang, Y. Jiang, D. Cun, W. Lin, Y. Fan and Y. Kawashima, 2003. Design of sustained-release nitrendipine microspheres having solid dispersion structure by quasi-emulsion solvent diffusion method. J. Controlled Release, 91: 375-384.
CrossRef - Di-Martino, P., C. Barthelemy, F. Piva, E. Joiris, G.F. Palmieri and S. Martelli, 1999. Improved dissolution behavior of fenbufen by spherical crystallization. Drug Dev., 25: 1073-1081.
Direct Link - Dunselman, P.H. and B. Edgar, 1991. Felodipine clinical pharmacokinetics. Clin. Pharmacokinet., 21: 418-430.
PubMed - Gupta, V.R., S. Mutalik, M.M. Patel and G.K. Jani, 2007. Spherical crystals of celecoxib to improve solubility, dissolution rate and micromeritic properties. Acta Pharm., 57: 173-184.
PubMed - Karavas, E., E. Georgarakis, D. Bikiaris, T. Thomas, V. Katsos and A. Xenakis, 2001. Hydrophilic matrices as a carriers in felodipine solid dispersion systems. Progr. Colloid Polym. Sci., 118: 149-152.
CrossRef - Kawashima, Y., F. Cui, H. Takeuchi, T. Niwa, T. Hino and K. Kiuchi, 1994. Improvements in flowability and compressibility of pharmaceutical crystals for direct tabletting by spherical crystallization with a two solvent system. Powder Technol., 78: 151-157.
CrossRef - Kawashima, Y., 1984. Development of spherical crystallization technique and its application to pharmaceutical system. Arch. Pharm. Res., 7: 145-151.
CrossRefDirect Link - Kawashima, Y., M. Okumura and H. Takenaka, 1982. Spherical crystallization: Direct spherical agglomeration of salicylic acid crystals during crystallization. Science, 216: 1127-1128.
CrossRefDirect Link - Lobenberg, R. and G.L. Amidon, 2000. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm., 50: 3-12.
CrossRefDirect Link - Saltiel, E., A.G. Ellrodt, J.P. Monk and M.S. Langley, 1988. Felodipine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in hypertension. Drugs, 36: 387-428.
PubMed - Sano, A., T. Kuriki, Y. Kawashima, H. Takeuchi, T. Hino and T. Niwa, 1992. Particle design of tolbutamide by spherical crystallization technique. V. Improvement of dissolution and bioavailability of direct compressed tablets prepared using tolbutamide agglomerated crystals. Chem. Pharm. Bull., 40: 3030-3035.
- Tapas, A.R., P.S. Kawtikwar and D.M. Sakarkar, 2009. Enhanced dissolution rate of felodipine using spherical agglomeration with Inutec SP1 by quasi emulsion solvent diffusion method. Res. Pharm. Sci., 4: 77-84.
Direct Link - Tapas, A.R., P.S. Kawtikwar and D.M. Sakarkar, 2010. Spherically agglomerated solid dispersions of valsartan to improve solubility, dissolution rate and micromeritic properties. Int. J. Drug Deliv., 2: 304-313.
CrossRefDirect Link - Usha, A.N., S. Mutalik, M.S. Reddy, A.K. Rajith, P. Kushtagi and N. Udupa, 2008. Preparation and in vitro, preclinical and clinical studies of aceclofenac spherical agglomerates. Eur. J. Pharm. Biopharm., 70: 674-683.
CrossRef - Vasconcelos, T., B. Sarmento and P. Costa, 2007. Solid dispersion as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discovery Toady, 12: 1068-1075.
CrossRef - Yadav, A.V. and V.B. Yadav, 2008. Designing of pharmaceuticals to improve physicochemical properties by spherical crystallization technique. J. Pharm. Res., 1: 105-112.
Direct Link - Yan, L., S. Jin and H. Zhong-Gui, 2009. In vitro dissolution behavior of felodipine tablets in different media. Chin. J. Pharma., 7: 425-430.
Direct Link - Yang, M.S., F. de Cui, B.G. You, Y.L. Fan, L. Wang, P. Yue and H. Yang, 2003. Preparation of sustained-release nitrendipine microspheres with Eudragit RS and Aerosil using quasi-emulsion solvent diffusion method. Int. J. Pharmac., 259: 103-113.
CrossRef