
Imam A.A. Mekkawy
Department of Zoology, Faculty of Science, Assiut University, Assiut, Egypt
LiveDNA: 20.3952
American Journal of Biochemistry and Molecular Biology
Year: 2017 | Volume: 7 | Issue: 1 | Page No.: 1-20







ABSTRACT
Background: The present study aimed at studying the phylogenetic relationships of 18 Lethrinus species and their associated trophic evolution on the basis of their filtered and validated DNA barcoding (cytochrome oxidase subunite I (COI)-sequences) released by the BOLD system database. Materials and Methods: About 98 COI sequences of 18 Lethrinus species were retrieved from BOLD system released nucleotide database and analyzed using different phylogenetic softwares. Results: Intra and inter-specific variations in COI-sequences were recorded with emphasis on geographic variations. Some COI-sequences were postulated to be of misidentified specimens and others were questionable leading to paraphyla. The COI-based phylogenetics using different statistical methods established two lineages in genus Lethrinus with L. haematopterus as ancestor group. These two lineages were corresponding with two obvious trophic types: Low-bodied species with conical lateral teeth and high bodied species with molariform teeth with the sister group L. haematopterus of high-bodied species with conical teeth. Conclusion: These findings emphasizes on the higher validity of DNA barcoding in discovering phylogenetics of genus Lethrinus in comparison to those based on cytochrome b.
PDF Abstract XML References Citation
Received: September 05, 2016;
Accepted: October 25, 2016;
Published: December 15, 2016
How to cite this article
Imam A.A. Mekkawy, 2017. Evolutionary Lineages in Genus Lethrinus (Family: Lethrinidae) and the Corresponding Trophic Evolution Based on DNA Barcoding. American Journal of Biochemistry and Molecular Biology, 7: 1-20.
DOI: 10.3923/ajbmb.2017.1.20
URL: https://scialert.net/abstract/?doi=ajbmb.2017.1.20
DOI: 10.3923/ajbmb.2017.1.20
URL: https://scialert.net/abstract/?doi=ajbmb.2017.1.20
INTRODUCTION
Lethrinids are taxonomically considered one of the most problematic group of tropical marine fish families. Their identification-related problems are primarily due to their relatively constant and conservative morphological and anatomical characteristics especially on the specific level1,2. The Lethrinidae has been classified with three other families into the superfamily Sparoidea in the phyletic sequence Nemipteridae, Lethrinidae and Sparidae plus Centracanthidae3,4. Different studies are in concern of the evolutionary relationships between these families3,5,6. Carpenter and Johnson3 placed on morphological bases, Nemipteridae as the sister group of Lethrinidae and Sparidae (including centracanthids).
According to Carpenter and Allen1, the Lethrinids are classified into two subfamilies: Lethrininae and Monotaxinae based on head scalation patterns and dorsal and anal-fin ray counts. Lo Galbo et al.2 reported that the Lethrininae includes Lethrinus with 29 species and the other four genera, Gnathodentex, Gymnocranius, Monotaxis and Wattsia comprise the Monotaxinae; all of monotaxine genera are monotypic except Gymnocranius. These researchers stated that for Lethrinus with very conservative morphological characters in particular, an independent data set is necessary to construct a phylogeny; cytochrome b gene was considered. Other researchers tried to assess some interspecific relationships in Lethrinus species based on scale morphology, morphometris, biology, isoenzymes and osteology in Egypt and Saudi Arabia7-10.
Piscivores, benthic invertebrate carnivores, zooplanktivores and herbivores are among the wide range of trophic types of sparoid fishes which are common components of hard bottom demersal fish communities with exception of Nemipterus3. The studies of evolution of the trophic types especially in the closely related fishes are rare2,11 and the reliable morphological characters in this concern are dentition and body shape, which are correlated with feeding type2,3,12. Carpenter12 studied the relationship of dentition, body shape and the associated specific feeding mode of species of Lethrinus with emphasize on their clues to the evolution of these feeding types. The Lethrinus species are demersal feeders with three distinct modes, low-bodied species with conical lateral teeth (LC), high bodied species with molariform (HM) teeth and high-bodied species with conical teeth (HC)2. According to researchers, the Lethrinus species with first two modes are mesocarnivore stalker and specialists, respectively and those with the latter mode are mesocarnivore generalists. Hanel and Sturmbauer11 hypothesized that the same trophic types evolved multiple time within the family. Lo Galbo et al.2 and Orrell et al.13 generated comprehensive phylogeny of sparidae genera and Lethrinus based on cytochrome b sequences and in concern with trophic types. Use of a more conservative gene may be necessary to adequate phylogeny of these genera.
Variable DNA sequence analyses have been reviewed14,15 and used for three decades to assist species identifications for different taxonomic groups in different laboratories16-23. This DNA-based approaches including DNA Barcoding (mitochondrial cytochrome c oxidase subunit I, COI) are used to identify fishes and resolve taxonomic problems including the discovery of new/cryptic species24-27. The DNA barcoding has been utilized to evaluate the incidence of fish species substitutions in North America28, Europe29 and Italy30,31. Due to its lower effective population size with rapid rate of evolution, various genes of mtDNA genome are applied to investigate different issues22,32-36. Meyer37 stated that different parts of the mitochondrial gene are known to evolve at different rates. The DNA barcoding methodology requires intraspecific DNA barcode variation substantially lesser than the interspecific one for accurate identification of individuals. One of the criticisms reported by many researchers including Lipscomb et al.38, Moritz and Cicero39 and Dasmahapatra and Mallet40 was that samples generally taken from a narrow geographic region may markedly lead to underestimated intraspecific level of variability.
Hebert et al.24 proposed the use of the mitochondrial DNA gene COI as a global bio-identification system for animals with empirical support extended from studies of invertebrates to birds19,41. Due to criticisms on such approach, Collins and Cruickshank42 referred to 7 deadly sins which are represent serious limitation to the utility of DNA barcoding especially in creating reference libraries. Becker43 also identified some of the human errors as the primary source of error in FISH-BOL barcode data. Spouge and Marino-Ramirez44 described a workflow for measuring the efficacy of DNA barcode in identifying species including the probability of correct identification.
According to the aforementioned findings, the present work aimed at determination of the evolutionary lineages in genus Lethrinus (Family: Lethrinidae) using their well-identified DNA barcodes (Cytochrome c oxidase subunit I, COI) recorded and released in the BOLD-system database and to examine the evolution of the three primary feeding types in Lethrinus species in comparison with the study of Lo Galbo et al.2 based on cytochrome b gene sequences. The 18 Lethrinus species used include L. lentjan, L. obsoletus, L. ornatus, L. crocineus, L. nebulosus, L. harak, L. mahsena, L. atkinsoni, L. laticaudis, L. semicinctus, L. ravus, L. conchyliatus, L. rubrioperculatus, L. microdon, L. xanthochilus, L. sp., L. olivaceus, L. sminiatus and L. haematopterus . Moreover, the origin of these species was also considered in their range of distribution.
MATERIALS AND METHODS
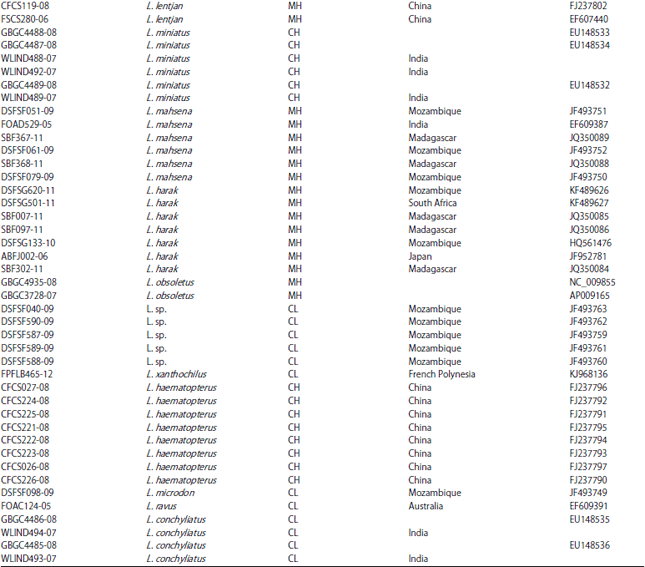
Data source: About 98 COI sequences of 18 Lethrinus species (Table 1) were retrieved from BOLD system released nucleotide database45. These Lethrinus species include L. lentjan, L. obsoletus, L. ornatus, L. crocineus, L. nebulosus, L. harak, L. mahsena, L. atkinsoni, L. laticaudis, L. semicinctus, L. ravus, L. conchyliatus, L. rubrioperculatus, L. microdon, L. xanthochilus, L. sp., L. olivaceus, L. miniatus and L. haematopterus. The L. sp., as referred in the database did not have a scientific name and is considered as it is. These species represent different geographical regions, India, Japan, Australia, China, Taiwan, Iran, South Africa, Madagascar and Mozambique. Moreover, different released sequences of different species of Gymnocranius (Lethrinidae), Acanthopagrus (Spariidae), Acanthurus (Acanthuridae) and Lophius (Lophiidae) from the same database were retrieved to work as outgroups in current analysis (Table 2). The specimen used in COI-sequence preparations were collected in different times and their sequences are published (BOLD system database).
Sequence and haplotype analyses: The COI-sequences of Lethrinus species in concern are analyzed using different softwares including MEGA 646 with Clustal W, Arlequin 3.547, DnaSP 5.1048, BioEdit 7.1.949 and SplitTree450. The outputs and parameters estimated by these softwares include aligned nucleotide sequences, Kimura 2-parameter (K2P) distances (overall mean, between mean and within mean species distances), phylogenetic trees and split tree, nucleotide composition, G+C content, numbers of polymorphic sites, parsimony informative sites, mutation and haplotypes, haplotype diversity, nucleotide diversity and average nucleotide difference (K).
| Table 1: | BOLD-system ID, GenBank_Accession ID collection locality and trophic catogeries for released DNA barcodes COI-sequences of different Lethrinus species used in the present study |
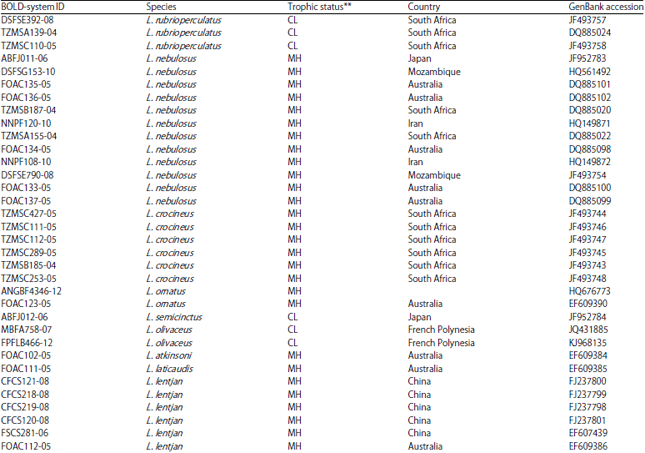  | |
**Dentition abbreviation, C: Conical, M: Molariform and submolariform, body types, H: High-bodied, L: Low-bodied | |
Source of variation parameters and statistics (FST, FSC and FCT) of analysis of molecular variance (AMOVA) approach were estimated by Arlequin 3.5. The default parameters of these programs were considered and applied.
Before subsequent phylogenetic analyses, BOLD-released COI-sequences of Lethrinus species were filtered using some of these softwares to exclude the irrelevant ones which led to paraphyla in this genus on the bases of sequences identity matrix 0.90 within each species using BioEdit 7.1.9. The status of these irrelevant sequences was verified.
For phylogenetic analyses and evolutionary history, Lethrinus species COI-sequences set (77 sequences of 18 species) are considered with each of three outgroups (Gymocranius, Gymnocranius+Acanthopagrus and Gymnocranius+Acanthopagrus+Acanthurus+Lophius) using three statistical methods: NJ, Neighbor Joining (Kimura 2-parameter method, Kimura, 1980); ML, Maximum Likelihood (Hasegama-kishino Yano/ Gamma distributed with invariant sites, G+I, Nearest-Neighbor-Interchange, NNI) and MP, Maximum Parsimony (Subtree-Pruning-Regrafting, SPR) with bootstrap replication 1000, 500 and 500 respectively in the units of the number of base substitutions per site.
| Table 2: | BOLD-system ID, GenBank_Accession ID and collection locality for released DNA barcodes COI-sequences of different species used as outgroups in the present study |
 | |
Codon positions included were 1st+2nd+3rd+noncoding. All positions containing gaps and missing data were eliminated from the dataset.
RESULTS
Sequence analysis: About 98 COI-sequences representing 18 Lethrinus species were considered in the current species released from BOLD SYSTEM database. These sequences representing different geographical regions including China, Australia, Japan, Taiwan, India, Iran, Madagascar, Mozambique and South Africa. The COI-sequences extended from 174-1548 bp with an average of 660±11.6. However, without extremes recorded in L. nebulosus and L. obsoletus, the other 94 COI-sequences representing the 18 species ranged from 590-682 with a mean of 649±1.64 bp. The NJ-based phylogenetic tree (Fig. 1) of all sequences (only 121 positions included in the final data set) exhibited paraphyletic and monophyletic species. The latter monophyletic groups include L. obsoletus, L. crocineus, L. harak and L. haematopterus. The remaining species are paraphyletics (Fig. 1). After, excluding the minicode sequence of 174 bp of L. nebulosus (NNPF178-10), the same situation of species paraphyla and monophyla with some variations is still recorded in spite of increased number of positions included in the final data set (527 bp) using different statistical methods (NJ, ML and MP). Accordingly, the within species variations and identity matrix in each Lethrinus species were examined by BioEdit program and in within each species sequences having identity less than 0.90 were excluded from subsequence analyses in an attempt to resolve the phylogenetic trees and the interspecific relationships.
Lethrinus nebulosus COI-sequences exhibited intraspecific variations including geographic ones. After exclusion of 5 sequences of L. nebulosus with Id<0.90, the overall mean distance (K2P) decreased from 0.185±0.014 for 16 sequences to 0.05±0.004 for sequences with Id 0.93. Moreover, the variable sites and parsimony informative sites reduced from 344-44 and from 57-40, respectively.
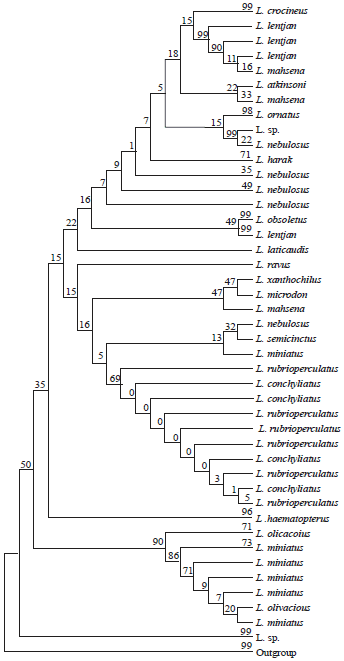 |
| Fig. 1: | Phylogenetic tree (based on the Neighbour Joining; Kimura-2 parameter) of the BOLD-system released 97 COI-sequences of 18 Lethrinus species with 6 COI-sequences of Gymnocranius species as outgroup (121 nucleotide positions are included in final data set, 1000 replications, bootstrapped) |
These sequences referred to two lineages: Japan-Australia and Mozambique-Iran; South Africa sequences appeared as sister group to these two lineages (Fig. 2a). However, the parsimony phylogenetic tree of all f sequences identifies the indian sequences as ancestor of the other sequences which reflects two different directions of L. nebulosus spreading (Fig. 2b): One towards the east (Australia-Japan-Taiwan) and the other towards the west (Iran-Mozambique-South Africa). However, in the subsequent PM and ML-based phylogenetic analyses of different final sets of Lethrinus species in concern, the two South African sequences of L. nebulosus (TZMSA155-04 and TZMSB187-04) were clustered as sister group to L. ornatus. So, inspite of their ID higher than 0.9, the specimen identification of these sequences as L. nebulosus is questionable.
Intraspecific variations of L. lentjan were also evident since its 11 COI-sequences are classified into two main lineages: China-Australia and Iran (Fig. 2c ). Iran sequences led to paraphyla in subsequent analyses. So, these sequences (Id<0.90) are excluded from the subsequent analyses of Lethrinus species. Accordingly, the overall mean distance decreased from 0.073±0.007 to 0.005±0.002.
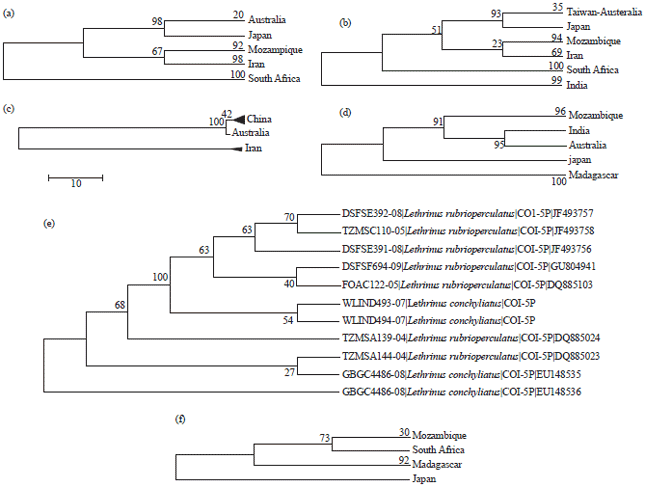 |
| Fig. 2(a-f): | Maximum parsimony-based phylogenetic trees (1000 replications) of the BOLD-system released COI-sequences of some Lethrinus species showing within species variations (a) L. nebulosus (only 12 COI sequence with ID>0.9, 639 position included in final data set with booting), (b) L. nebulosus (all 16 COI sequence; 534 position included in final data set with booting), (c) L. lentjan (11 COI sequences; 641 position included in final data set), (d) L. mahsena (8 coi sequences; 651 position included in final data set), (e) L. rubrioperculatus and L. conchyliatus (552 position included in final data set) and (f) L. harak (7 COI sequences; 650 position included in final data set) |
Moreover, the polymorphic sites changed from 87-8 with high reductions in other parameters such as haplotype and nucleotide diversities and average number of nucleotide differences.
Lethrinus atkinsoni sequence (FOAC 102) of BOLD-system 98 released COI-sequences is included with L. mahsena sequence set since all preliminary analyses of its phylogenetics emphasized on its affiliation to L. mahsena not to L. atkinsoni (Fig. 2d). After the removal of Japanese sequences (ABFJ022-06) of L. mahsena with Id<0.90, the overall mean distance slightly reduced from 0.083±0.008 to 0.057±0.007 with variations in other parameters. Lethrinus mahsena sequence set exhibited geographic variations in Mozambique, Madagascar, India, Australia and Japan regions. The excluded Japanese sequence is clustered with Madagascar sequences in the booted phylogenetic parsimony tree.
One of L. rubrioperculatus sequences (TZMSC144) with Id>0.90 is excluded. So, the overall mean distance reduced from 0.863±0.118 to 0.002±0.001. However, on clustering, these sequences interfere in the subsequent analyses only with the L. conchyliatus sequences in spite of their Id matrix 0.94 up to 1 (Fig. 2e). Accordingly, other three L. rubrioperculatus sequences (DSF391, DSF694 and FOAC122) are excluded to prevent paraphyla in subsequent analyses.
The FOAC117-05 sequence with Id<0.90 is excluded from L. miniatus set leading to little change in the overall mean distance from 0.053±0.005 to 0.002±0.001. Six sequence set of L. miniatus (Id>0.99) separated as a single unite in all subsequent analyses. Lethrinus olivaceus sequences leading to paraphyla in L. miniatus and L. olivaceus complex are also excluded in final analyses of Lethrinus species. The DSFSE398 and FOAC126 sequences of L. olivaceus may belong to L. miniatus since these sequences are separated within L. miniatus set.
Except for DSFSE390 sequence with Id<0.90, all L. sp. sequences (Id>0.99) are separated as one unite in all subsequent phylogenetic analyses by different methods. Also, each of the sequence sets of L. concineus, L. harak, L. haematopterus and L. obsoletus is separated as one unit with no paraphyla under different methods; their Id matrices have very high values. The majority of these species exhibited intraspecific variations including geographic ones. In L. harak sequence set (Id>0.95 up to 0.998), there are two lineages: Mozambique-South Africa and Madagascar; Japanese sequence represent an ancestor to these lineages (Fig. 2f). In L. haematopterus sequence set, phylogenetics refer to two main groups in the same locality under different statistical methods and analyses. Each of the other six Lethrinus species is only represented by one sequence.
According to the aforementioned inspection and analysis of Bold-system released COI-data of Lethrinus species, only 77 sequences out of 98 ones are included in the subsequent phylogenetic analyses. Outgroups used in these analyses are represented by some sequences of Gymnocranius (Family: Lethrinidae), Acanthopagrus (Family: Spariidae, a sister group to Lethrinidae), Lophius (Family: Lophiidae) and/or Acanthurus (Family: Acanthuridae).
The overall mean distance for 77-sequences set of 18 Lethrinus species is 0.140±0.011 whereas these distances with Gymocranius outgroup and Lethrinus-all outgroups set are 0.149±0.011 and 0.182±0.011, respectively. The within mean group distance of Lethrinus-all outgroups set is 0.009±0.003 with range of 0.00-0.059, nearly the same for Lethrinus species only (0.009±0.005). The between group mean distance of Lethrinus species is 0.153±0.003 (0.005-0.214) whereas those between Lethrinus species and each of Gymnocranius, Acanthopagrus, Acanthurus and Lophius outgroups are 0.211±0.002 (0.193-0.231), 0.230±0.002 (0.186-0.262), 0.228±0.003 (0.143-0.259) and 0.227±0.003 (0.206-0.254). The total between group mean distance for Lethrinus-all outgroup set is 0.193±0.003 (0.005-0.266).
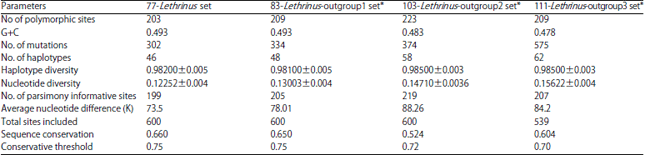
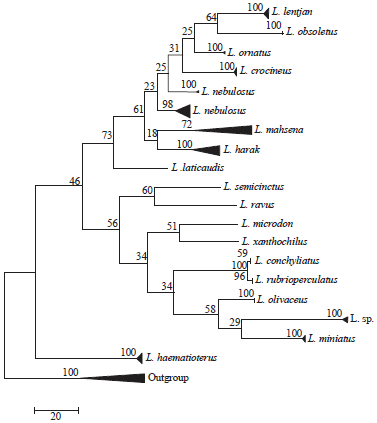
For phylogenetic analyses, Lethrinus species COI-sequences set (77 sequences of 18 species) are considered with each of three outgroups (Gymocranius, Gymnocranius+Acanthopagrus and Gymnocranius+ Acanthopagrus+Acanthurus+Lophius) using three statistical methods: NJ (K2P), ML (Hasegama-kishino Yano/G+I) and MP (Subtree-Pruning-Regrafting) with bootstrap replication of 1000, 500 and 500, respectively. The population parameters of these four sets used are given in Table 3. In all cases, there are two lineages of Lethrinus species (Fig. 3-5). The first lineage (Lineage 1) includes L. lentjan, L. obsoletus, L. ornatus, L. crocineus, L. nebulosus, L. harak, L. mahsena and L. laticaudis and the second one (Lineage 2) comprises L. semicinctus, L. ravus, L. conchyliatus, L. rubrioperculatus, L. microdon, L. xanthochilus, L. sp., L. olivaceus, L. miniatus and L. haematopterus acts as a sister group to the two lineages in the majority of cases of the three statistical methods used for the three sets of data with different outgroups. The minimum evolution tree not mentioned here, also emphasized on such conclusion. However, in the booted PM of 83-set and NJ of 103-set, L. haematopterus act as a sister group to lineage 1. Also, this species act as a sister group to lineage 2 only by unbooted MP analysis of 103-set. As a results, one can postulated L. haematopterus as an ancestor to the two COI-based lineages of Lethrinus recorded.
Lethrinus conchyliatus and L. rubrioperculatus sequences are clustered together in all cases. The following groupings of species are recorded to be constant in all analyses with different methods: L. lentjan+L. obsoletus, L. harak+L. mahsena, L. sp.+L. olivaceus+L. miniatus, L. semicinctus+L. ravus and L. microdon+L. xanthochilus. These interspecific genetic relationships reflect in the majority of cases, the corresponding morphological ones in taxonomic case.
| Table 3: | Some population parameters of the four COI-sets used in the final COI-based phylogenetic analyses of 18 Lethrinus species in concern |
 | |
*Outgroup 1: Gymocranius, outgroup 2: Gymnocranius+Acanthopagrus and outgroup 3: Gymnocranius+Acanthopagrus+Acanthurus+Lophius | |
 |
| Fig. 3: | Maximum parsimony-based phylogenetic tree of the BOLD-system released 77 COI-sequences of 18 Lethrinus species with 6 COI-sequences of Gymnocranius species as outgroup (600 nucleotide positions are included in final data set, 500 replications) |
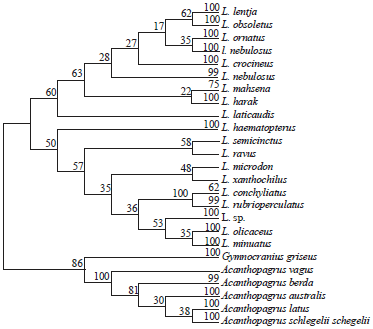 |
| Fig. 4: | Maximum parsimony-based phylogenetic tree of the BOLD-system released 77 COI-sequences of 18 Lethrinus species with 26 COI-sequences of Gymnocranius (6) and Acanthopagrus (20) species as outgroup (600 nucleotide positions are included in final data set, 500 replication, bootstrapped) |
Except for NJ analyses of 83-set and 103-set, L. nebulosus sequences are distributed between two groups in all cases, one represents the majority of its sequences and the other one includes only the two South African sequences which are clustered as sister group to L. ornatus. In lineage 1, L. laticaudis is found to be ancestor to the remaining species in the majority of cases. However, in NJ and booted ML of 111-set, L. mahsena resolves as sister to lineage 1.
 |
| Fig. 5: | Maximum parsimony-based phylogenetic tree of the BOLD-system released 77 COI-sequences of 18 Lethrinus species with 34 COI-sequences of Gymnocranius (6), Acanthopagrus (20), Acanthurius (4) and Lophius (4) species as outgroup (539 nucleotide positions are included in final data set, 500 replication) |
These latter rare cases may be due to inclusion of the outgroup species away of Lethrinidae and Sparidae (the lethrinid sister group) and the corresponding bootstrapping.
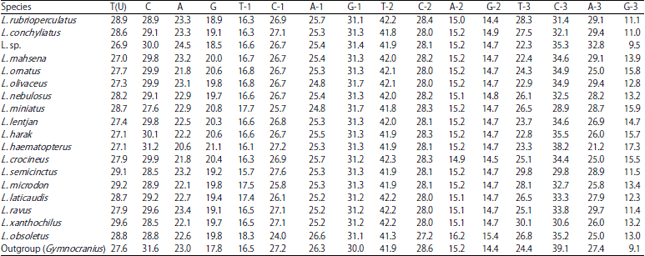
Haplotype and AMOVA analyses: Out of 600 nucleotide positions of 83-set included in the analysis, 209 are found to be variable with 205 parsimony informative sites, G+C of 0.443, mutations of 334, haplotypes of 48, high haplotype diversity of 0.981±0.005, nucleotide diversity of 0.13003±0.004 and average nucleotide difference of 78.01. The average nucleotide compositions of the 83-set species are given in Table 4. The distribution of the 48 haplotypes (Fig. 6, 7) between species is given in Table 5; no sharing in haplotypes between species is recorded. The tree of these haplotypes on statistical bases of NJ and MP reflects a pattern of haplotype grouping corresponding to that of tree-based sequences since two lineages were identified with haplotypes of L. haematopterus as sister group to the two lineages; haplotypes of Gymnocranius sequences were found to be a sister group to Lethrinus species considered. The genetic distance (K2P) between haplotypes ranged between 0.005-1.52 with an average of 0.600±0.009. Also, p-distance averaged 0.347±0.004 with a range of 0.005-0.563.
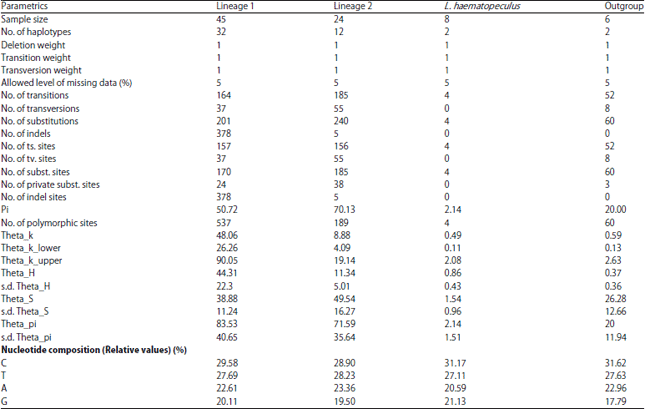
As reflect by phylogenetic analyses, the 83-sequence set of the 18 Lethrinus species considered with the outgroup, Gymnocranius species are classified into four groups (clades) for further analysis by AMOVA (Fig. 3, Table 6). These groups include sequences of species of lineage 1 (L1), species of lineage 2 (L2), L. haematopterus (LH) and the outgroup. The between group average distances (K2P) of the first Lethrinus three groups (L1 and L2 = 0.117±0.014, L1 and LH = 0.177±0.016 and L2 and LH = 0.186±0.016) were greater than their within group average distances (L1, L2 and LH = 0.087±0.007, 0.121±0.010 and 0.004±0.002, respectively).
| Table 4: | Average nucleotide compositions of COI-sequences of Lethrinus species and outgroup in concern |
 | |
| Table 5: | Distribution of the 48 COI-based haplotypes among Lethrinus species and outgroup in concern |
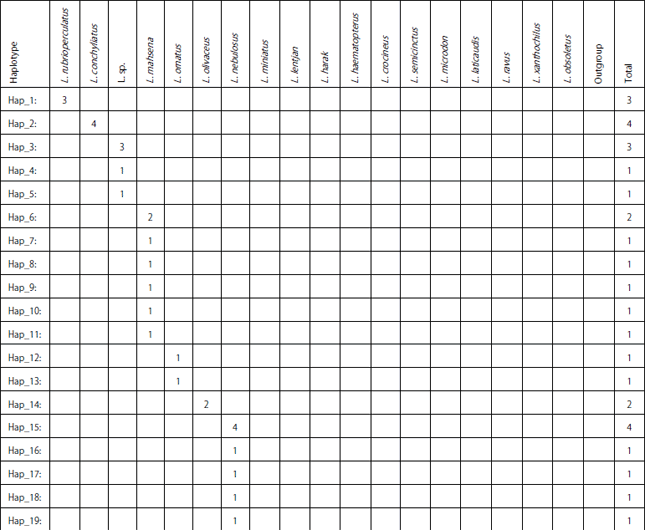 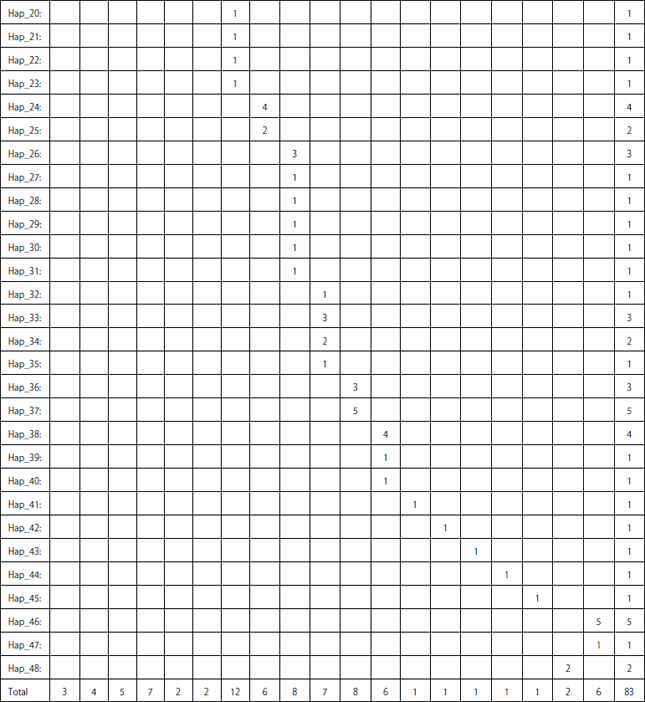 | |
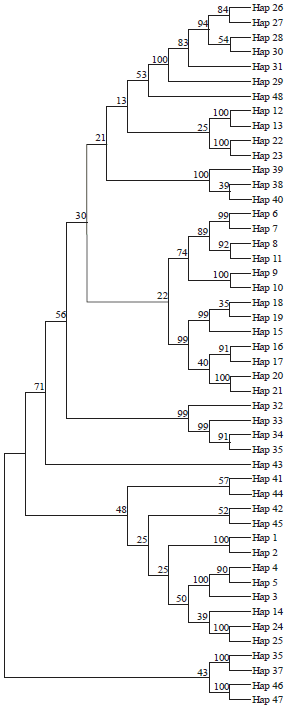 |
| Fig. 6: | Maximum parsimony-based phylogenetic tree of the 48 haplotypes of the BOLD-system released 77 COI-sequences of 18 Lethrinus species with 6 COI-sequences of Gymnocranius species (208 nucleotide positions are included in final data set, 1000 replications) |
| Table 6: | AMOVA analysis of the four groups (Lineage 1, lineage 2, L. haematopterus and the outgroup, Gaymncranius species) of the 83-COI sequence set of Lethrinus species studied |
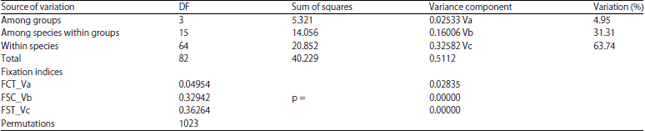 | |
The AMOVA statistics (FST, FSC and FCT) are significant with p>0.00000, 0.00000 and 0.02835, respectively (Table 6). These findings referrers to significance between the four clades identified in 83-sequence set as well as between species within lineages. Overall average FST value referred to great genetic differentiation among species. Individual FST matrix between species at significance of 0.05 is given in Table 7. Also, the variability in genetic structure characteristics of the four clades or groups is given in Table 8.
The two COI-based lineages identified in current Lethrinus species phylogenetics are found to be correlated with the two dentition-body patterns (HM and LC) with no overlapping except for L. miniatus (a mesocarnivore generalist, HC which is clustered with L. olivaceus (CL) and L. sp. (CL) as subclade in lineage 2 (the mesocarnivore stalkers, CL) in all analyses based on different statistical methods. L. haematopterus (HC) in the majority of cases as postulated previously resolves as the most ancestral species and positions sister to all remaining Lethrinus species.
| Table 7: | Computing conventional Lethrinus pairwise FSTs matrix from COI-haplotype frequencies and matrix of significance values in AMOVA at 0.05 level, permutations = 110 |
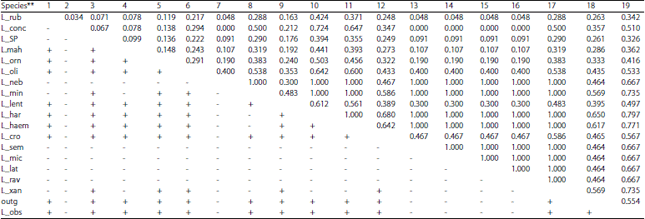 | |
**Species abbreviations include the first three or four letters of the species name, outg: Outgroup, Gymnocranius species | |
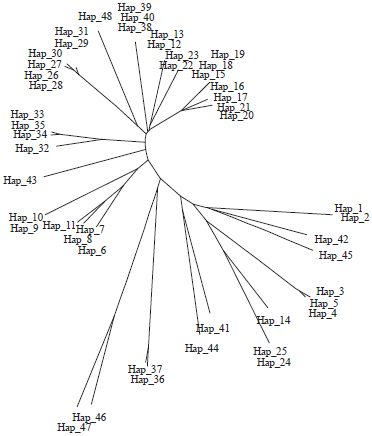 |
| Fig. 7: | Splitstree phylogenetic tree of the 48 haplotypes of the BOLD-system released 77 COI-sequences of 18 Lethrinus species with 6 COI-sequences of Gymnocranius species |
| Table 8: | Genetic structural characteristics of the four clades or groups identified on the bases of phylogenetic analyses of the 83-COI-set of the 18 Lethrinus species in concern with Gymnocranius sequences as outgroup |
 | |
In addition, L. laticaudis (HM) positions sister to the remaining species in lineage 1 (mesocarnivore specialist, HM) in the majority of cases of current phylogenetic analyses.
DISCUSSION
The DNA barcoding is a well established method for specimen identification and species discovery using a short standardized region (648 bp, the folmer region) of DNA24,42,51,52. Potentially, this regions contain enough information to resolve 10-100 million species24. However, misconceptions pervading the DNA barcoding literature were considered and highlighted by Collins and Cruickshank42 in terms of 7 deadly sins of DNA barcoding with suggestions of possible improvements. Some of these suggestions and those of Lv et al.53 were applied to the analysis of the 97 Lethrinus species sequences in concern and about 20 sequences were excluded to prevent paraphyla and to build resolved phylogenetic tree of Lethrinus species considered. Some of these sequences represented inadequate a priori identification of specimens and their possible correct identification was suggested according to liberal tree-base method53. Accordingly, one can suggest that GenBank and BOLD system database sequences must be accepted for phylogenetic analysis after their filtering and validity analysis to avoid the deadly sins of DNA and misconception and hence transformation of morphology-based taxonomic problems to these databases.
Hajibabaei and McKenna54 referred to difficulty of getting a full length DNA barcode in older preserved museum specimens and in processed biological materials such as food products, pharmaceuticals and nutraceuticals. So, DNA mini-barcodes have been recovered with effectiveness in biodiversity analysis and in specimen identification to a species level in museum samples54-56. In the present study, NJ-based phylogenetic tree of all sequences (only 121 positions included in the final data set due to sequence (174 bp) of L. nebulosus specimen (NNPF178-10) exhibited paraphyletic and monophyletic species. After removal of the inadequate sequences except that of NNPF178-10, paraphyla disappeared and the pattern of Lethrinus two lineages with the sister group, L. haematopterus (postulated later with 600 postion included) was not resolved since 137 positions are included in the final data set. This finding referred to the little information reflected by mini-codes.
Some researchers and investigators applied more than one statistical method and model2 in phylogenetic studies and others used only one method57. These statistical methods are built on different assumptions and produced minor variable outputs especially on the interspecific relationships. Bootstrapping also exhibits such minor variability. These minor variations do not prevent the resolution of phylogenetic trees in sequence data freed of inadequate ones prior to phylogenetic analyses. Such situation was evident in the present study since the outputs of the majority of method and models referred to similar pattern of COI-based Lethrinus interspecific relationship.
The three trophic categories, LC, HC and HM recorded in emperor fishes, genus Lethrinus in relation to body form and dentition type were morphologically, genetically and evolutionary considered by different researchers2,3,12. Carpenter and Allen1 and Carpenter12 described the variations in the body-feeding-habitat characteristics of these tropical and sub-tropical fishes which distributed mostly in the Indo-West Pacific. Al-Sufiani7 referred to the head region of some Lethrinus species to be morphologically important in their differention by truss characteristics. Other researchers including Alexander58 referred to general trends in the evolution of body shape in relation to swimming behavior, habitat and feeding types. However, using morphology in inferring trophic type evolution of Lethrinus species is problematic since these morphological characters are typically correlated with feeding mode and are potentially homoplasious2. These researchers used mitochondrial DNA sequences (cytochrome b) to determine phylogenetic hypothesis for Lethrinus, not related to trophic morphological characteristics. The results of Lo Galbo et al.2 clearly inferred a monophyletic Lethrinus with two well defined lineages within Lethrinus exhibiting two distinct trophic types (LC and HM)) as delineated by Carpenter12. In the present study, similarly these two lineages were well identified on the basis of DNA barcoding of COI with different in their ancestor (sister group). These findings as postulated by Lo Galbo et al.2 indicated the primary radiation with Lethrinus occurred separately thin these two lineages. In contrast, the same trophic type evolved separately several times in the more speciose Sparidae11,13.
Using cytochrome b, Lo Galbo et al.2 recorded L. miniatus (HC) as a well-supported Lethrinus clade aside from the two primary trophic groups, working as basal species, sister to all other Lethrinus species. On the other hand, in the COI-based present work L. haematopterus (LC) was found to be a well-supported Lethrinus clade aside from the two trophic lineages and then ancestor to these lineages. The author of the current work phylogenetically (PM and NJ methods, 500 and 1000 replications, respectively) analyzed 34 cytochrome b sequences (1140 bp) of 20 Lethrinus species (Table 9) retrieved from NCBI database59,60. It is postulated that L. miniatus did not position as ancestor of that genus, only separated with L. erythropterus in one cluster in one of Lethrinus lineages (Fig. 8).
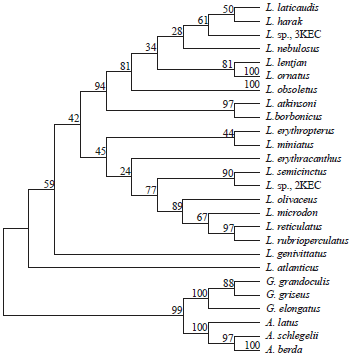 |
| Fig. 8: | Maximum parsimony-based phylogenetic tree of 24 cytochrome b sequences (1140 bp) of 20 Lethrinus species with 10 other sequences (1140 bp) of 6 species of genera: Gymnocranius and Acanthopagrus as outgroup (1137 nucleotide positions are included in final data set, 500 replications, booted). Sequences retrieved from NCBI databases |
| Table 9: | List of cytochrome b sequences of Lethrinus species and outgroups used in their phylogenetic tree retrieved from GenBank database |
 | |
The differences between the four categories: L. haematopterus, the other two lineages and the out groups (Gymnocranius) were emphasized to be significant by AMOVA analyses of their haplotypes. The phylogenetic analyses of the corresponding 48 haplotypes reflect also the same phylogenetic-based pattern of relationship between these lineages and their ancestor. According to the results based on cytochrome b and COI, which species of L. haematopterus and L. maniatus, one can postulated as ancestor to Lethrinus species. In the present study, L. maniatus was only clustered with L. olivaceus (LC) and L. sp. (LC) in all analyses using different methods. So, L. haematopterus will be considered as the ancestor of Lethrinus species especially the DNA barcode, COI is recommended as a well-established method by many researchers in the evolutionary and phylogenetic studies as well as in global market for fisheries and aquaculture products24,25,28,51,53,61,62.
The Lethrinus genus, Gymnocranius with HC-trophic type positions in all analysis as a sister group of genus Lethrinus in the present study. Generally one can emphasized on hypothesis of Lo Galbo et al.2 that the two primary trophic types (HM and LC) are evolved from a high-bodied conical-toothed (HC) ancestor. The placement of L. miniatus in lineage 2 (CL-lineage) of the present study may be explained as a support of this hypothesis. In the study of Lo Galbo et al.2, L. erythropterus (HM) and L. erythracanthus (HC) were grouped within LC-lineage whereas L. genivittatus (LC) and L. atlanticus (HC) were clustered within HM-lineage. These four Lethrinus species are not represented in the current COI-database. It is expected that such overlapping of these species could not be recorded if they are represented in COI-database. These findings of L. haematopterus (only restricted to temperate waters in East Asia) and L. miniatus with antitropical distribution1 referred to a temperate water form to be the ancestor of the genus Lethrinus2. Finally, on the basis of aforementioned discussion, do genetic analysis of different mitochondrial genes and/or of different genes of the whole genome lead to different gene-based ancestors or one one genome-based ancestor to a given genus Like Lethrinus? A question needs to be answered.
CONCLUSION
According to the current study, one can concluded the following points: (1) DNA sequences databases and sources must be validated and filtered to prevent paraphyla in phylogenetic studies and to correct misidentification of specimens, (2) During construction of phylogenetic trees, different statistical methods should be considered, (3) Based on COI sequences released from BOLD system database, genus Lethrinus exhibited two lineages with L. haematopterus as ancestor, (4) This taxonomic status of Lethrinus was in accordance to the three trophic types (LC, HM and HC) and (5) COI-based analyses is moe better than those based on cytochrome b and mini-DNA barcoding.
SIGNIFICANCE STATEMENT
The current study is important due to:
| • | The taxonomic morphometric-related problems in Lethrinus |
| • | The appearing of DNA barcoding as a valid new technology in solving phylogenetic problems |
| • | The application of this technology on Lethrinus species for determination of evolutionary trends in corresponding of trophic evolution |
REFERENCES
- Carpenter, K.E. and G.R. Allen, 1989. FAO species catalogue, Vol. 9. Emperor fishes and large-eye breams of the world (Family Lethrinidae): An annotated and illustrated catalogue of lethrinid species known to date. FAO Fisheries Synopsis 125, United Nations Development Programme, Food and Agriculture Organization of the United Nations, Rome, Italy.
- Lo Galbo, A.M., K.E. Carpenter and D.L. Reed, 2002. Evolution of trophic types in emperor fishes (Lethrinus, Lethrinidae, Percoidei) based on cytochrome b gene sequence variation. J. Mol. Evol., 54: 754-762.
CrossRefDirect Link - Carpenter, K.E. and G.D. Johnson, 2002. A phylogeny of sparoid fishes (Perciformes, Percoidei) based on morphology. Ichthyol. Res., 49: 114-127.
CrossRefDirect Link - Johnson, D.G., 1993. Percomorph phylogeny: Progress and problems. Bull. Mar. Sci., 52: 3-28.
Direct Link - Hanel, R. and C. Sturmbauer, 2000. Multiple recurrent evolution of trophic types in Northeastern Atlantic and Mediterranean seabreams (Sparidae, Percoidei). J. Mol. Evol., 50: 276-283.
Direct Link - Orrell, T.M., K.E. Carpenter, J.A. Musick and J.E. Graves, 2002. Phylogenetic and biogeographic analysis of the Sparidae (Perciformes: Percoidei) from cytochrome b sequences. Copeia, 3: 618-631.
Direct Link - Bhattacharya, M., A.R. Sharma, B.C. Patra, G. Sharma and E.M. Seo et al., 2016. DNA barcoding to fishes: Current status and future directions. Mitochondrial DNA Part A, 27: 2744-2752.
CrossRefDirect Link - Trivedi, S., A.A. Aloufi, A.A. Ansari and S.K. Ghosh, 2016. Role of DNA barcoding in marine biodiversity assessment and conservation: An update. Saudi J. Biol. Sci., 23: 161-171.
CrossRefDirect Link - Barrett, R.D.H. and P.D.N. Hebert, 2005. Identifying spiders through DNA barcodes. Can. J. Zool., 83: 481-491.
CrossRefDirect Link - Clare, E.L., B.K. Lim, M.D. Engstrom, J.L. Eger and P.D. Hebert, 2007. DNA barcoding of neotropical bats: Species identification and discovery within Guyana. Mol. Ecol. Notes, 7: 184-190.
CrossRefDirect Link - Costa, F.O., J.R. DeWaard, J. Boutillier, S. Ratnasingham, R.T. Dooh, M. Hajibabaei and P.D. Hebert, 2007. Biological identifications through DNA barcodes: The case of the Crustacea. Can. J. Fish. Aquat. Sci., 64: 272-295.
CrossRefDirect Link - Hebert, P.D.N., E.H. Penton, J.M. Burns, D.H. Janzen and W. Hallwachs, 2004. Ten species in one: DNA barcoding reveals cryptic species in the Neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A., 101: 14812-14817.
CrossRefPubMedDirect Link - Hebert, P.D., M.Y. Stoeckle, T.S. Zemlak and C.M. Francis, 2004. Identification of birds through DNA barcodes. PLoS Biol., Vol. 2.
CrossRefDirect Link - Smith, M.A., B.L. Fisher and P.D.N. Hebert, 2005. DNA barcoding for effective biodiversity assessment of a hyperdiverse arthropod group: The ants of Madagascar. Philos. Trans. R. Soc. London B: Biol. Sci., 360: 1825-1834.
CrossRefDirect Link - Ward, R.D., B.H. Holmes, W.T. White and P.R. Last, 2008. DNA barcoding Australasian chondrichthyans: Results and potential uses in conservation. Mar. Freshwater Res., 59: 57-71.
CrossRefDirect Link - Ward, R.D., T.S. Zemlak, B.H. Innes, P.R. Last and P.D.N. Hebert, 2005. DNA barcoding Australia's fish species. Philos. Trans. R. Soc. B: Biol. Sci., 360: 1847-1857.
CrossRefPubMedDirect Link - Hebert, P.D.N., A. Cywinska, S.L. Ball and J.R. deWaard, 2003. Biological identifications through DNA barcodes. Proc. R. Soc. B: Biol. Sci., 270: 313-321.
CrossRefPubMedDirect Link - Kim, S., H.S. Eo, H. Koo, J.K. Choi and W. Kim, 2010. DNA barcode-based molecular identification system for fish species. Mol. Cells, 30: 507-512.
CrossRefDirect Link - Lakra, W.S., M.S. Verma, M. Goswami, K.K. Lal and V. Mohinda et al., 2011. DNA barcoding Indian marine fishes. Mol. Ecol. Resour., 11: 60-71.
CrossRefPubMedDirect Link - Winterbottom, R., R. Hanner, M. Burridge and M. Zur, 2014. A cornucopia of cryptic species-a DNA barcode analysis of the gobiid fish genus Trimma (Percomorpha, Gobiiformes). ZooKeys, 381: 79-111.
CrossRefDirect Link - Wong, E.H.K. and R.H. Hanner, 2008. DNA barcoding detects market substitution in North American seafood. Food Res. Int., 41: 828-837.
CrossRefDirect Link - Miller, D.D. and S. Mariani, 2010. Smoke, mirrors and mislabeled cod: Poor transparency in the European seafood industry. Frontiers Ecol. Environ., 8: 517-521.
CrossRefDirect Link - Barbuto, M., A. Galimberti, E. Ferri, M. Labra, R. Malandra, P. Galli and M. Casiraghi, 2010. DNA barcoding reveals fraudulent substitutions in shark seafood products: The Italian case of palombo (Mustelus spp.). Food Res. Int., 43: 376-381.
CrossRefDirect Link - Filonzi, L., S. Chiesa, M. Vaghi and F.N. Marzano, 2010. Molecular barcoding reveals mislabelling of commercial fish products in Italy. Food Res. Int., 43: 1383-1388.
CrossRefDirect Link - Apostolidis, A.P., D. Loukovitis and C.S. Tsigenopoulos, 2008. Genetic characterization of brown trout (Salmo trutta) populations from the Southern Balkans using mtDNA sequencing and RFLP analysis. Hydrobiologia, 600: 169-176.
CrossRefDirect Link - He, A., Y. Luo, H. Yang, L. Liu, S. Li and C. Wang, 2011. Complete mitochondrial DNA sequences of the Nile tilapia (Oreochromis niloticus) and Blue tilapia (Oreochromis aureus): Genome characterization and phylogeny applications. Mol. Biol. Rep., 38: 2015-2021.
CrossRefDirect Link - Mandal, A., V. Mohindra, R.K. Singh, P. Punia, A.K. Singh and K.K. Lal, 2012. Mitochondrial DNA variation in natural populations of endangered Indian feather-back fish, Chitala chitala. Mol. Biol. Rep., 39: 1765-1775.
CrossRefDirect Link - Diaz, J., G.V. Villanova, F. Brancolini, F. del Pazo, V.M. Posner, A. Grimberg and S.E. Arranz, 2016. First DNA barcode reference library for the identification of south american freshwater fish from the lower Parana river. PloS One, Vol. 11.
CrossRefDirect Link - Shen, Y., L. Guan, D. Wang and X. Gan, 2016. DNA barcoding and evaluation of genetic diversity in Cyprinidae fish in the midstream of the Yangtze river. Ecol. Evol., 6: 2702-2713.
CrossRefDirect Link - Lipscomb, D., N. Platnick and Q. Wheeler, 2003. The intellectual content of taxonomy: A comment on DNA taxonomy. Trends Ecol. Evol., 18: 65-66.
CrossRefDirect Link - Moritz, C. and C. Cicero, 2004. DNA barcoding: Promise and pitfalls. PLoS Biol., Vol. 2.
CrossRefDirect Link - Dasmahapatra, K.K. and J. Mallet, 2006. DNA barcodes: Recent successes and future prospects. Heredity, 97: 254-255.
CrossRefDirect Link - Hogg, I.D. and P.D.N. Hebert, 2004. Biological identification of springtails (Hexapoda: Collembola) from the Canadian Arctic, using mitochondrial DNA barcodes. Can. J. Zool., 82: 749-754.
CrossRefDirect Link - Collins, R.A. and R.H. Cruickshank, 2013. The seven deadly sins of DNA barcoding. Mol. Ecol. Resour., 13: 969-975.
CrossRefDirect Link - Becker, S., R. Hanner and D. Steinke, 2011. Five years of FISH-BOL: Brief status report. Mitochondrial DNA, 22: 3-9.
CrossRefDirect Link - Spouge, J.L. and L. Marino-Ramirez, 2012. The practical evaluation of DNA barcode efficacy. Methods Mol. Biol., 858: 365-377.
CrossRefDirect Link - Ratnasingham, S. and P.D.N. Hebert, 2007. BOLD: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes, 7: 355-364.
CrossRefDirect Link - Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform., 5: 150-163.
CrossRefPubMedDirect Link - Excoffier, L., G. Laval and S. Schneider, 2005. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online, 1: 47-50.
PubMedDirect Link - Librado, P. and J. Rozas, 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25: 1451-1452.
CrossRefPubMedDirect Link - Hall, T.A., 1999. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acid Symp. Ser., 41: 95-98.
Direct Link - Huson, D.H. and D. Bryant, 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol., 23: 254-267.
CrossRefDirect Link - Little, D.P., 2011. DNA barcode sequence identification incorporating taxonomic hierarchy and within taxon variability. PLoS ONE, Vol. 6.
CrossRefDirect Link - Schindel, D.E. and S.E. Miller, 2005. DNA barcoding a useful tool for taxonomists. Nature, 435: 17-17.
CrossRefPubMedDirect Link - Lv, J., S. Wu, Y. Zhang, Y. Chen and C. Feng et al., 2014. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasites Vectors, Vol. 7.
CrossRefDirect Link - Meusnier, I., G.A.C. Singer, J.F. Landry, D.A. Hickey, P.D.N. Hebert and M. Hajibabaei, 2008. A universal DNA mini-barcode for biodiversity analysis. BMC Genomics, Vol. 9.
CrossRefDirect Link - Srirama, R., B.R. Gurumurthy, U. Senthilkumar, G. Ravikanth, R.U. Shaanker and M.B. Shivanna, 2014. Are mini DNA-barcodes sufficiently informative to resolve species identities? An in silico analysis using Phyllanthus. J. Genet., 93: 823-829.
CrossRefDirect Link - John, A., C. Prasannakuma, P.S. Lyla, S.A. Khan and K.C.A. Jalal, 2010. DNA barcoding of Lates calcarifer (Bloch, 1970). Res. J. Biol. Sci., 5: 414-419.
CrossRefDirect Link - Benson, D.A., I. Karsch-Mizrachi, D.J. Lipman, J. Ostell and E.W. Sayers, 2009. GenBank. Nucleic Acids Res., 37: D26-D31.
CrossRefPubMedDirect Link - Wheeler, D.L., T. Barrett, D.A. Benson, S.H. Bryant and K. Canese et al., 2007. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 35: D5-D12.
CrossRefDirect Link - Pradhan, V., Y. Kamble, V. Ladniya and M. Mogul, 2015. A overview of species identification by DNA barcoding. Int. J. Curr. Microbiol. Applied Sci., 4: 127-140.
Direct Link - Wong, L.L., E. Peatman, J. Lu, H. Kucuktas and S. He et al., 2011. DNA barcoding of catfish: Species authentication and phylogenetic assessment. PLoS ONE, Vol. 6.
CrossRefDirect Link