
Xinyan Leng
Department of Microbiology, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, Jiangsu, People`s Republic of China
Rongmei Fei
Department of Microbiology, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing 210095, Jiangsu, People`s Republic of China
Asian Journal of Animal and Veterinary Advances
Year: 2012 | Volume: 7 | Issue: 12 | Page No.: 1301-1311







ABSTRACT
White spot syndrome is a viral infection of penaeid shrimps which is highly infectious and lethal, terminating shrimps rapidly. Outbreaks of this disease can wipe out entire shrimp populations within a few days. To reduce the mortality of shrimps and increase the production of marine industry, the research focused on the changes on the virus’s proteins. The research is aimed at the molecular reconstruction of one of the important proteins from the White Spot Syndrome Virus (WSSV). By exploring the unique DNA sequences as well as the corresponding protein, the initial target shall be achieved soon. According to the published gene sequence of White Spot Syndrome Virus (WSSV), a pair of specific primers were designed. Using an isolate of WSSV collected in Ganyu, Jiangsu Province, China, as template, a gene fragment of VP36A was amplified by Polymerase Chain Reaction (PCR). The PCR product was firstly TA-cloned into a pMD-18T vector and then inserted into a pET-28a plasmid to create a recombinant plasmid and finally transformed into the host strain BL21. The recombinant protein was expressed in the form of inclusion bodies with 1.0 mmol L-1 Isopropyl-β-D-thiogalactopyranoside(IPTG) at 37°C. According to the results of an Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), the recombinant protein had the expected size of 36 kDa. The purified recombinant protein was tested in a Western blot to confirm that the target protein had been successfully expressed. This showed that the special DNA in WSSV can be replicated and the protein can be expressed by a very cheap and sufficient way in a very limiting time, which will be utilized in the further experiments related to the functional identification of protein (VP36A).
PDF Abstract XML References Citation
Received: August 13, 2012;
Accepted: November 03, 2012;
Published: November 19, 2012
How to cite this article
Xinyan Leng and Rongmei Fei, 2012. Cloning, Expression and Purification of Recombinant Envelope Protein VP36A of White Spot Syndrome Virus. Asian Journal of Animal and Veterinary Advances, 7: 1301-1311.
DOI: 10.3923/ajava.2012.1301.1311
URL: https://scialert.net/abstract/?doi=ajava.2012.1301.1311
DOI: 10.3923/ajava.2012.1301.1311
URL: https://scialert.net/abstract/?doi=ajava.2012.1301.1311
INTRODUCTION
In 1992, reports from Taiwan showed the first epidemic outbreak due to White Spot Syndrome Virus (WSSV) (Chen, 1995), followed by losses reported from China in 1993 (Huang et al., 1995), where it led to significant losses of the shrimp farming industry. From then on, outbreaks expanded to territorial waters in Japan and Korea also in 1993 and then shrimp farming in East and South Asia were severely affected. In late 1995, the disease was reported in the U.S. and 1998 in Central and South America, followed by European countries. So far, the disease has been present in shrimp-growing regions all over the world in addition to Australia. White spot syndrome virus can severely infect a wide range of hosts, such as farmed and wild shrimp and wild crabs and also lead to a high level of mortality up to 100% (Lo et al., 1996). Thus, the disease has caused a virtual collapse in global shrimp fishery as well as of shrimp farms and hatcheries (Flegel, 1997; Tsai and Huang, 1995), which can also threaten the balance of entire marine ecosystems (Jory, 1999). Transmission of the virus occurs mainly through oral ingestion and water-borne routes in farms (horizontal transmission) (Kanchanaphum et al., 1998) or from infected mother prawns in the case of shrimp hatcheries (vertical transmission) (Huang and Song, 1999).
WSSV is the only member of the family Nimaviridae that discovered so far. It is symbolized as a large, rod-shaped, double-stranded DNA virus (Nadala Jr. et al., 1998). It has an outer lipid bilayer membrane envelope and cannot form an inclusion body during its infectious period (Mayo, 2002). WSSV genomic DNA was purified from infected prawns in 1997 and was fully sequenced in 2001. Nearly 180 Open Reading Frames (ORFs) of 50 or more amino acids have been found within the complete virus genome (Yang et al., 1997; Van Hulten et al., 2001; Yang et al., 2001). Recently, studies of the pathogenicity of WSSV are mostly focusing on viral genes such as the function of viral proteins. The transcription and translation of WSSV-VP36A are conducted by genes in the ORF WSSV134, encoding a total of 297 amino acids. The theoretical molecular weight of VP36A is 33.1 kDa, while a dimensional electrophoresis profile revealed a weight of 36 kDa. VP36A is one of the membrane proteins that functions in the early phase of infection and the usage of its antibody can significantly delay the initial infection in Procambarus shrimps (Li et al., 2006a). Envelope proteins play an important role during the processes of adsorption, invasion, packaging and release of enveloped viruses like WSSV (Chiu and Chang, 2002; Hsiao et al., 1999; Lin et al., 2000; Chazal and Gerlier, 2003). VP36A also contains a Arginine-Glycine-Aspartic acid (RGD) locus (Tsai et al., 2004), was justified by analyzing the function of VP36A-related factors during the viral infectious progress. In addition, the recombined envelope protein will provide advantageous experimental material with high purity and quantity, which would be needed in further research about the role of VP36A in initial infection (Li et al., 2006b).
In the present study, the T7-RNA polymerase based pET system was employed to express the viral envelope protein in Escherichia coli. The ORF-WSSV134 fragment was cloned into a pET-28a (+) vector, in which the genes of interest were cloned downstream to the E. coli thioredoxin (TRX) chimera in order to increase the solubility of the target protein and tagged with an N-terminal His tag to allow easy purification (Rosenberg et al., 1996). The recombinant protein was then purified with a purification column. In addition, SDS-PAGE and Western blot were applied in order to verify the obtained protein. Therefore, a foundation is established to discern the functional mechanisms between WSSV envelope proteins, such as VP36A and shrimp cells in future studies.
MATERIALS AND METHODS
This research was initiated in 2011. The WSSV strain used in the experiments was obtained from Ganyu, Jiangsu Province in 2011 and kept in the Microbiology and Immunology Laboratory, College of Veterinary Medicine, Nanjing Agricultural University, China. E. coli strain DH5-a competent cells and BL21 (DE3) competent cells were purchased from Tiangen. Restriction endonucleases EcoRI and XholI, pMD18-T Simple Vector and T4 DNA ligase were obtained from Takara. T7 polymerase expression vector pET-28a (+) was stored in the laboratory. A PCR-product purification kit, DNA purification kit and agarose gel DNA purification kit were purchased from Geneaid, while a 2xTaqMaster Mix was purchased from Dongsheng Biotech. A protein purification kit (HisTrapTM HP) was purchased from GE Healthcare, HRP secondary antibodies were from Boster Biotech. 3,3’,5,5’-Tetramethylbenzidine (TMB) was from Tiangen. Other reagents were imported or domestic analytical reagents.
DNA purification: Gills of contagious shrimps stored at –40°C were ground in Phosphate Buffer Sline (PBS). The liquid was frozen at –20°C for 2 h and thoroughly thawed at room temperature. This freeze-thaw move was repeated twice. Afterwards, the grinding liquid was centrifuged in a 2 mL EP tube for 10 min at 13,000 rpm (Sambrook and Russell, 2001). Following the instructions of the Viral Nucleic Acid Extraction Kit II manual (Geneaid), total DNA was extracted from the supernatant of the centrifuged liquid.
Cloning of the VP36A gene: Primers were designed by software Primer 5.0 according to the cDNA sequences of WSSV ORF 134 within white spot syndrome virus Thailand strain (GenBank accession No.: AF369029) and were synthesized by Invitrogen. VP36A was amplified by using the following primers:
Forward: 5'-GGAATTCGCATTACAGGAAAAGGATAT- 3'
Reverse: 5'-GGCCTCGAGAAACTACTACTATACATATT-3'
Containing an EcoRI (forward, underlined) and an XholI site (reverse, underlined), respectively. The following PCR protocol was used: pre-denaturation at 94°C for 5 min; denaturation at 94°C for 30 sec annealing at 51°C for 60 sec, extension at 72°C for 30 sec, 30 cycles, followed by an extension at 72°C for 10 min. The PCR product was verified by 1% Agarose Gel Electrophoresis (AGE) and then extracted by a Gel/PCR DNA Fragments Extraction kit (Geneaid) following the manual’s instructions.
Construction of recombined plasmid pET- VP36A: After purification from the agarose gel, the amplified fragment was ligated into a pMD18-T vector to produce pMD-VP36A. E. coli DH5-a was transformed with the ligative mixture and grown at 37°C after plating on an LB agar medium containing 100 μg mL-1 ampicillin. Positive clones were selected and identified with PCR and restriction enzyme digestion. DNA sequences were verified by Invitrogen Biotechnology. The TA-cloned pMD-VP36A was digested with EcoRI/XholI and followed by ligation with EcoRI/Xhol-digested pET-28a (+) to produce pET-VP36A. The expression vector was transformed into E. coli BL21 (DE3). The positive clones were identified by PCR and restriction enzyme digestion and their DNA sequences were verified by Invitrogen Biotech.
Expression of VP36A: The E. coli BL21(DE3) harboring pET-P36A were inoculated at a proportion of 1:100 into LB containing 100 μg mL-1 kanamycin (1%), then incubated at 37°C with shaking overnight. The overnight culture was inoculated into LB with 100 μg mL-1 kanamycin (1%) for continuous amplification until the OD600 value reached about 0.6 (which took less than 3 h). After instantly adding IPTG at a final concentration of 1 mmol L-1, the culture was incubated for another 4 h to induce the expression of the target protein. Before and during the period of inducement, 1 mL of the bacterium culture was collected every hour. After high speed centrifugation of the collected cultured liquid, an SDS-PAGE was performed to identify the optimum time point for the expression of the target protein. The empty vector pET-28a (+) cultured at the same conditions was treated as control.
Examination of the expression site in bacteria: To obtain sufficient quantity of bacteria for further analysis, more than 400 mL of LB with 1% 100 μg mL-1 kanamycin was used in culturing E. coli BL21 (DE3) that carried pET-P36A at the optimum induced expression. The cultured liquid was collected for high-speed centrifugation at 10,000 g for 10 min at 4°C, with the supernatant being discarded while the sediment was washed three times with sterile PBS. The bacteria were resuspended in the same PBS and then sonicated (at 200 W for 4 sec work time and 8 sec interval) until the resuspension became more transparent. The supernatant and sediment were collected after a centrifugation of sonicated resuspension at 4°C for 20 min at 10,000 g. After an SDS-PAGE analysis of both supernatant and sediment, the VP36A protein was shown to be expressed either in the form of solution or inclusion bodies.
Purification inclusion bodies and renaturation of recombined protein: After verifying the expression of VP36A in the inclusion bodies, the sediment was washed by inclusion body purgation buffer and centrifuged at 10,000 g at 4°C for 10 min. The sediment was resuspended with binding buffer in a 30°C water bath for 1 h and the resuspension was centrifuged at 10,000 g at 4°C for 10 min. The supernatant was filtered by a 0.45 μm membrane and then purified by a His trap affinity column (GE Healthcare). The purified protein was dialyzed by PBST containing urea of a concentration subsequently decreasing from 6 to 4 to 2 to 0 M. After a thorough dialysis the protein solution was collected and stored at 4°C.
Verification by Western blot analysis: The renatured protein was analyzed by SDS-PAGE to isolate the fused protein. The gel was then subjected to Western blotting; proteins were transferred onto nitrocellulose membranes using a semi-dry electroblot apparatus (Bio-Rad) in electroblotting buffer (25 mM Tris, 190 mM glycine, 20% methanol) at a constant voltage of 100 V for 2 h. The membrane was immersed in blocking buffer (PBST containing 5% (w/v) skimmed milk) at 4°C overnight, followed by incubation with polyclonal mouse anti (His)’-HRP (1:3000) (Invitrogen) for 2.5 h in a shaker (50 rpm, 37°C). The membrane was washed three times with PBST for 5 min each at 37°C with gentle rotation. The HRP secondary antibody was then added and incubated at 37°C in a shaker for 1 h. The membrane was again washed as described above. Subsequently, detection was performed with chromogenic substrate TMB (4,4'-bi-2,6-xylidine, Tiangen) solution.
RESULTS
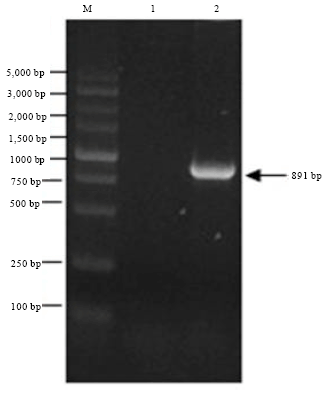
Cloning of the WSSV-VP36A gene: Using the DNA of WSSV as template, the products of PCR were separated by 1.5% agarose gel electrophoresis. PCR amplification of the WSSV-VP36A gene yielded an 891 bp DNA fragment with the expected sequence (Fig. 1).
Construction and identification of recombinant vectors: The fragments of WSSV-VP36A and pMD18-T vector were ligated to a plasmid pMD-VP36A, which was identified by PCR (the same primers used in amplifying the template). The results showed a band between 750 and 1000 bp as expected (Fig. 2). The plasmids pET-28a (+) and pMD-VP36A were digested with EcoRI and XholI, ligated by T4 DNA ligase and then transformed by E. coli DH5-a. The recombinants were selected and identified by PCR and double digestion.
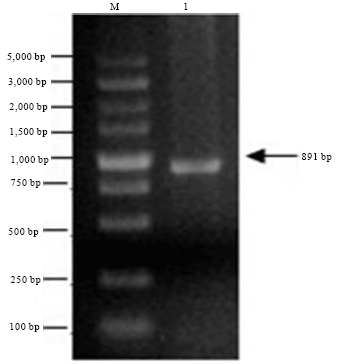 | |
| Fig. 1: | PCR products of VP36A gene. M: DNA marker, Lane 1: PCR products of gene VP36A (891 bp) |
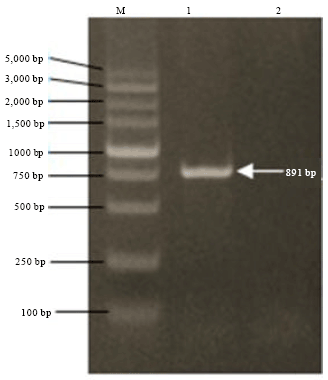 | |
| Fig. 2: | Identification of recombinant plasmid pMD-VP36A by PCR, M: DNA marker, Lane 1: pMD-VP36A plasmid used as template (891 bp), Lane 2: Blank control sample |
The expected bands were verified through AGE (Fig. 3, 4). The sequencing results revealed that neither pre-termination nor frame shift had occurred and that the sequence matched the complete genome in GenBank, which indicates the successful recombined plasmid construction.
 | |
| Fig. 3: | Identification of recombinant plasmid pET-VP36A by PCR, M: DNA marker, Lane 1: pET-VP36A plasmid used as template (891 bp), Lane 2: Blank control sample |
 | |
| Fig. 4: | Identification of recombinant plasmid pET-VP36A by restriction enzyme, M: DNA marker, Lane 1: pET-VP36A digested with EcoRI and XholI (4.9+0.89 kb) |
Expression of recombined plasmid and renaturation of inclusion bodies: The expression of recombined plasmid was detected after inducing the pET-VP36A that transformed into competent cell E. coli BL21 (DE3) by IPTG (final concentration 1 mM L-1) for 6 h.
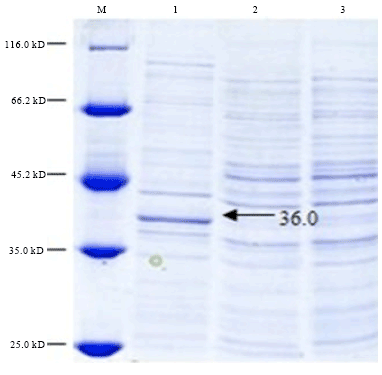 | |
| Fig. 5: | Analysis of recombinant protein pET-VP36A by SDS-PAGE, M: Molecular weight protein marker, Lane 1: pET-VP36A induced by IPTG for 6 h, Lane 2: pET-28a (+) induced by IPTG for 6 h, Lane 3: pET-28a (+) before induction |
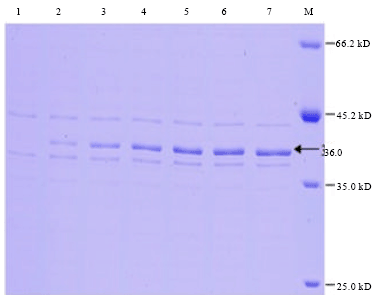 | |
| Fig. 6: | Analysis of recombinant protein pET-VP36A induced at different times by SDS-PAGE, M: Molecular weight protein marker, Lane 1: pET-VP36A before induction, Lanes 2-7: pET-VP36A induced by IPTG for 1, 2, 3, 4, 5, 6 h, respectively |
The weight of the expressed protein was between 35.0 and 45.2 kDa, thus, matching the expected weight of around 36.0 kDa (Fig. 5, 6). Both the supernatant and sediment of centrifuged sonicated bacteria were analyzed in an SDS-PAGE assay. The results showed that the fused protein was virtually expressed in the inclusion body and was almost absent in the supernatant (Fig. 7). After being purified by urea-containing PBST, the WSSV envelope protein VP36A was analyzed by SDS-PAGE. The resulting band of around 36 kDa size indicated that the target protein was obtained (Fig. 8).
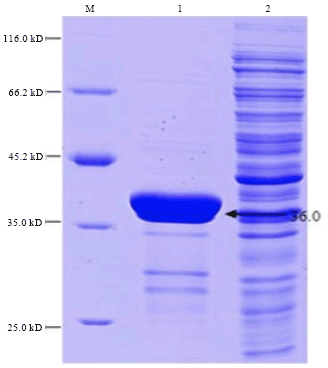 | |
| Fig. 7: | Analysis bacteria lysates of recombinant protein by SDS-PAGE, M: Molecular weight protein marker, Lane 1: Precipitation of bacteria with recombinant pET-VP36A after ultrasonic treatment, Lane 2: Supernatant of bacteria with recombinant pET-VP36A following ultrasonic treatment |
 | |
| Fig. 8: | Analysis of purified recombinant protein pET-VP36A by SDS-PAGE, M: Molecular weight protein marker, Lane 1: Purified recombinant protein analysis |
Western blot of fused protein: After renaturation, the recombined protein was analyzed by Western blot to identify the expression level of the fused protein. The result suggested a high level of protein expression as well as the identification of WSSV-VP36A (Fig. 9).
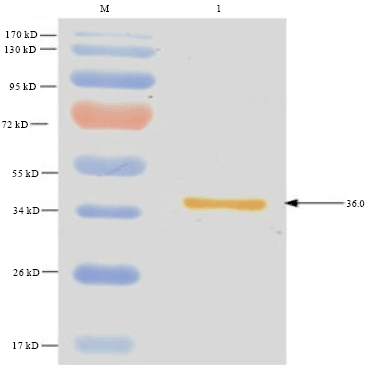 | |
| Fig. 9: | Western blot analysis, M: Molecular weight protein marker, Lane 1: Western blot analysis |
DISCUSSION
Previous studies of WSSV have mainly focused on the tissues and range of viral hosts as well as on DNA replication. Van Hulten et al. (2001) and Yang et al. (1997) completed the sequencing of the complete genome of WSSV (Van Hulten et al., 2001; Yang et al., 2001). In recent years, the focus of WSSV-related research has shifted to infection mechanisms and pathogenesis, including viral proteomics (Durand et al., 1997; Wang et al., 2000). More than 40 kinds of proteins have been reported (Escobedo-Bonilla et al., 2008), including structural proteins such as VP28, VP26 and VP35 that are necessary during the formation of a morphologically mature contagious viral particle. Other identified proteins include non-structural proteins like thymidine kinase, thymidine kinase and ribonucleotide reductase, which play a regulatory role during viral replication.
In this study, the non-structural protein VP36A was studied and the pET-28a (+) plasmid of the series of pET-TRX fused expression plasmids was used in the experiment. The Trx tag, which could enhance the solubility and stability of exogenous proteins expressed in E. coli, has been introduced into pET-TRX plasmids as a fusion partner. Attached to the plasmid was the protein purification tag His tag that is conducive to the purification of recombinant proteins and contains a T7 RNA polymerase-binding site. Recurring to the T7 RNA polymerase on host bacteria strain E. coli BL21 and the plasmid, the target gene can be transcribed and expressed. This method of transcription has the advantage of being easy to control and selective (Xu, 2003). Furthermore, fused expression could reduce the destabilization of protein products in E. coli and improve the expression levels of target genes and the antigenicity of expressed proteins (Novagen, 2010). As a result, the extraneous gene was expressed efficiently and steadily during the experiment.
To design the PCR primers, both the restriction sites and protective bases should be taken into consideration. EcoRI and XholI were chosen because they can be easily obtained and are relatively cheap. They also both possess an optimal digestion temperature of around 37°C, so, that synchronous digestion can be realized and efficiency can be improved. In addition, protective bases added to the 5’-end (G added for EcoRI and GGC for XholI) can significantly influence the binding of endonucleases to DNA duplexes and enzyme digestion of DNA. Throughout the experiment, the designed primers acted with excellent specificity that yielded large quantities of PCR amplification products. The SDS-PAGE also showed a protein of expected size. The expression of the protein in the inclusion body rather than in the supernatant of the recombinant plasmid after IPTG inducement suggested high yields of protein products, which was also verified by Western blot.
WSSV is mainly spread between shrimps by vertical infection through the intake of virus hosts. This characteristic of viral spread has been related to the capsule, as the nucleocapsid of non-capsule WSSV is not infectious. This was demonstrated by Li et al. (2006b) who performed a neutralization test with VP36A-specific antibodies (Li et al., 2006b).
CONCLUSION
The results showed that the antibody could delay the initial WSSV infection of Procambarus clarkii, although the mortality of the experimental animals was still 100% 11 days post injection. Although, WSSV-VP36A is a non-structural protein, it still actively functions during infection, which is probably realized by its reciprocity with other proteins. In this study, the target protein was successfully expressed and purified, which should enable further in-depth research on protein interactions, including structural and nonstructural proteins as well as envelope and nucleocapsid proteins.
ACKNOWLEDGMENTS
We are grateful to the Laboratory of Microbiology in College of Veterinary Medicine, Nanjing Agricultural University for providing the experimental equipments in need. This work was supported by grants from the Special Fund for Agro-scientific Research in the Public Interest (No. 201103034) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
REFERENCES
- Chazal, N. and D. Gerlier, 2003. Virus entry, assembly, budding and membrane rafts. Microbiol. Mol. Biol. Rev., 67: 226-237.
CrossRef - Chiu, W.L. and W. Chang, 2002. Vaccinia virus J1R protein: A viral membrane protein that is essential for virion morphogenesis. J. Virol., 76: 9575-9587.
PubMedDirect Link - Durand, S., D.V. Lightner, R.M. Redman and J.R. Bonami, 1997. Ultrastructure and morphogenesis of White Spot Syndrome Baculovirus (WSSV). Dis. Aquat. Organ., 29: 205-211.
Direct Link - Escobedo-Bonilla, C.M., V. Alday- Sanz, M. Wille, P. Sorgeloos, M.B. Pensaert and H.J. Nauwynck, 2008. A review on the morphology, molecular characterization, morphologenesis and pathogenesis of white spot syndrome virus. J. Fish. Dis., 31: 1-18.
PubMed - Flegel, T.W., 1997. Major viral diseases of the black tiger prawn (Penaeus monodon) in Thailand. World J. Microbiol. Biotechnol., 13: 433-442.
CrossRef - Hsiao, J.C., C.S. Chung, W. Chang, 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol., 73: 8750-8761.
PubMed - Huang, J., X.L. Song, J. Yu and C.H. Yang, 1995. Baculoviral hypodermal and hematopoietic necrosis: Study on the pathogen and pathology of the explosive epidemic disease of shrimp. Mar. Fish. Res., 16: 1-10.
Direct Link - Li, L., S. Lin and F. Yang, 2006. Characterization of an envelope protein (VP110) of White spot syndrome virus. J. Gen. Virol., 87: 1909-1915.
CrossRef - Li, L.J., J.F. Yuan, C.A. Cai, W.G. Gu and Z.L. Shi, 2006. Multiple envelope proteins are involved in white spot syndrome virus (WSSV) infection in crayfish. Arch. Virol., 151: 1309-1317.
CrossRef - Lin, C.L., C.S. Chung, H.G. Heine and W. Chang, 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection In vitro and In vivo. J. Virol., 74: 3353-3365.
Direct Link - Lo, C.F., C.H. HO, S.E. Peng, C.H. Chen and H.C. Hsu et al , 1996. White spot syndrome baculovirus (WSBV) detected in cultured and captured shrimp, crabs and other arthropods. Dis. Aquat Org., 27: 215-225.
CrossRefDirect Link - Huang, C.C. and Y.L. Song, 1999. Maternal transmission of immunity to White Spot syndrome Associated Virus (WSSV) in shrimp (Penaeus monodon). Dev. Comp. Immunol., 23: 545-552.
CrossRef - Kanchanaphum, P., C. Wongteerasupaya, N. Sitidilokratana, V. Boonsaeng and S. Panyim et al., 1998. Experimental transmission of White-Spot Syndrome Virus (WSSV) from crabs to shrimp Penaeus monodon. Dis. Aquat. Org., 34: 1-7.
PubMed - Nadala Jr., F.C., L.M. Tapay and P.C. Loh, 1998. Characterization of a non-occluded baculovirus-like agent pathogenic to penaeid shrimp. Dis. Aquat. Org., 33: 221-229.
PubMed - Mayo, M.A., 2002. A summary of taxonomic changes recently approved by ICTV. Arch. Virol., 147: 1655-1663.
CrossRefPubMedDirect Link - Sambrook, J. and D.W. Russell, 2001. Molecular Cloning: A Laboratory Manual. 3rd Edn., Cold Spring Harbor Laboratory Press, New York, USA., ISBN-13: 9780879695774, Pages: 2344.
Direct Link - Tsai, J.M., H.C. Wang, J.H. Leu, H.H. Hsiao, A.H.J. Wang, G.H. Kou and C.F. Lo, 2004. Genomic and proteomic analysis of thirty-nine structural proteins of shrimp white spot syndrome virus. J. Virol., 78: 11360-11370.
CrossRefDirect Link - Van Hulten, M.C.W., J. Witteveldt, S. Peters, N. Kloosterboer, R. Tarchini, M. Fiers, H. Sandbrink et al., 2001. The white spot syndrome virus DNA genome sequence. Virol., 286: 7-22.
CrossRefDirect Link - Wang, Q., B.T. Poulos and D.V. Lightner, 2000. Protein analysis of geographic isolates of shrimp white spot syndrome virus. Arch. Virol., 145: 263-274.
Direct Link - Yang, F., J. He, X. Lin, Q. Li, D. Pan, X. Zhang and X. Xu, 2001. Complete genome sequence of the shrimp white spot bacilliform virus. J. Virol., 75: 118-121.
CrossRefDirect Link - Yang, F., W. Wang, R. Chen and X. Xu, 1997. A simple and efficient method for purification of prawn baculovirus DNA. J. Virol. Meth., 67: 1-4.
CrossRefPubMedDirect Link