
R.E. Kartasasmita
School of Pharmacy, Institut Teknologi Bandung, Jalan Ganesha 10, Bandung, 40132, Indonesia
R. Herowati
School of Pharmacy, Institut Teknologi Bandung, Jalan Ganesha 10, Bandung, 40132, Indonesia
T. Gusdinar
School of Pharmacy, Institut Teknologi Bandung, Jalan Ganesha 10, Bandung, 40132, Indonesia
Journal of Applied Sciences
Year: 2010 | Volume: 10 | Issue: 23 | Page No.: 3098-3104







ABSTRACT
Some flavonoids, including quercetin, were reported to show inhibitory activities against inducible Nitric Oxide Synthase (iNOS), an isoenzyme responsible for nitric oxide formation. The objectives of this research are to obtain binding and inhibitory parameters of some quercetin derivatives on iNOS by means of docking method and prediction of their oral absorption and distribution properties. Seven selected flavonoids and ten quercetin derivatives were used as ligands for the study. The iNOS structure was obtained from Brookhaven protein databank (PDB ID: 1M9T) and docking study was performed using ArgusLab free software. Binding energy (ΔGbind) and hydrogen bond were used to analyze interactions between ligands and active site of iNOS. The PreADMET on line program was used to predict the oral absorption and distribution properties including permeability for Caco-2 cell, HIA (human intestinal absorption) and plasma protein binding. The results showed that hydrogen bonds formed between flavonoids and iNOS always involved the amide groups of glycine (A365) and trypsine (A366) residues in iNOS active site and 4-carbonyl group of flavonoids. In the docked form, the planar region of A-ring of flavonoids oriented to the heme plane of iNOS. Thus the 4-carbonyl group and planar region of A-ring of flavonoids are essential for the binding with iNOS. Linear regression of binding energy versus negative logarithm of IC50 of flavonoids gave an equation of -log IC50 = -0.399 ΔGbind- 5.608 (R2 = 0.686; p<0.01) and predicted IC50 of Quercetin was 76.79 μM. The human intestinal absorption (HIA) and Caco-2 cell permeability values of quercetin were 63.5% and 3.4 nm sec-1 while its plasma protein binding was 93.2%, respectively. Quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetate showed lower predicted IC50 and better absorption and distribution properties than quercetin.
PDF Abstract XML References Citation
Received: June 22, 2010;
Accepted: September 28, 2010;
Published: October 14, 2010
How to cite this article
R.E. Kartasasmita, R. Herowati and T. Gusdinar, 2010. Docking Study of Quercetin Derivatives on Inducible Nitric Oxide Synthase and Prediction of their Absorption and Distribution Properties. Journal of Applied Sciences, 10: 3098-3104.
DOI: 10.3923/jas.2010.3098.3104
URL: https://scialert.net/abstract/?doi=jas.2010.3098.3104
DOI: 10.3923/jas.2010.3098.3104
URL: https://scialert.net/abstract/?doi=jas.2010.3098.3104
INTRODUCTION
Nitric Oxide (NO) is produced from L-arginine in mammalian tissue by Nitric Oxide Synthase (NOS) enzymes. There are three NOS isoenzymes, i.e., nNOS (constitutive in neuronal tissue), eNOS (constitutive in vascular endothelial cells) and iNOS (inducible by cytokine in macrophages and hepatocytes) (Knowles and Moncada, 1994). Constitutively expressed eNOS and nNOS are responsible for low physiological levels of NO, whereas larger amounts of NO are produced by iNOS. iNOS is induced by microbial products, such as lipopolysaccharide (LPS) and inflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α) and interferon-γ (INF-γ) in macrophages and some other cells (Hamalainen et al., 2007). NO production is increased in respon to inflammatory stimuli and mediates the destructive effects (Korhonen et al., 2005). Because of the importance of NO derived from iNOS in inflammation response, there were several research efforts to find a selective iNOS inhibitor. The compounds inhibiting expression or activity of iNOS are proposed to be potential as anti-inflammatory agents (Knowles and Moncada, 1994).
Flavonoids (Fig. 1) are a group of naturally occurring polyphenolic compounds containing two benzene rings linked together with a heterocyclic pyran or pyrone ring and ubiquitously found in fruits and vegetables.
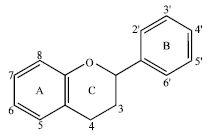 | |
| Fig. 1: | General structure of flavonoid |
| Table 1: | Structures and in vitro inhibitory activities of flavonoids and their predicted binding energy to iNOS |
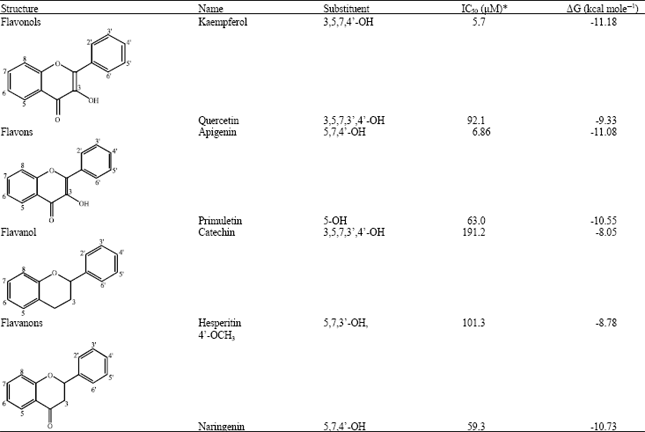 | |
| *IC50 values were taken from Olszanecki et al. (2002) | |
Flavonoids have been known to show potential health benefits (Hamalainen et al., 2007). Some of flavonoids have been reported to have anti-inflammatory properties by inhibition of iNOS in culture medium of LPS-stimulated, i.e., kaempferol, quercetin, apigenin, primuletin, catechin, hesperetin and naringenin (Table 1) (Olszanecki et al., 2002). Quercetin (3,3’,4’,5,7-pentahydroxyflavon) is a flavonoid suitable to be chosen as the lead compound for development of safe anti-inflammatory agent, because in addition to its anti-inflammatory effect, quercetin also shows protective effect in gastrointestinal track (Morikawa et al., 2003; Coskun et al., 2004). However, clinical use or clinical using of quercetin was limited by its low oral bioavailability (Peng et al., 2008). Thus, molecular modification of quercetin was needed to increase the oral bioavailability and enhance its pharmacological properties.
Molecular docking is a tool in structural molecular biology and structure-based drug discovery. The goal of ligand-protein docking is to understand and predict molecular recognition, finding likely binding modes and predicting binding affinity (Morris and Lim-Wilby, 2008). ArgusLab 4.0.1. is a freely available docking software, which serves two docking engine, i.e., GADock and ArgusDock (ArgusLab, 2004). These engines are capable for binding free energy calculation between proteins and ligands. Furthermore, ArgusLab is easy and inexpensive program useful for virtual screening (Oda and Takahashi, 2009).
Prediction of ADME (absorption, distribution, metabolism and excretion) properties has been developed to reduce the probability of the failure at the development stage of drug candidates. PreADMET is a web-based application for predicting ADME data and building drug-like library using in silico method. This program is useful for the construction of drug absorption prediction system. In absorption, it provides prediction models for in vitro Caco-2 -cell and MDCK cell (Madin-Darby canine kidney) assay as well as in silico HIA (human intestinal absorption). In distribution, it provides prediction of plasma protein binding and BBB (blood brain barrier) penetration (Lee et al., 2003). The objectives of this research are to obtain binding and inhibitory parameters of some quercetin derivatives on iNOS by means of docking method and prediction of their oral absorption and distribution properties.
MATERIALS AND METHODS
All research activities were conducted in Laboratory of Drug Design and High Computing, School of Pharmacy ITB, Indonesia.
Structural models: Structure coordinate for iNOS was taken from the RCSB Protein Data Bank (PDB ID: 1M9T), in which inducible NOS oxygenase domains was cocrystallized with 3-bromo-7-nitroindazole (Rosenfeld et al., 2002). The 3D structures of flavonoids and quercetin derivatives were first constructed using Arguslab 4.0.1., then were optimized using Austin Model 1 (AM1), 200 maximum iteration, followed by conjugate gradient minimization to a Root Mean Square (RMS) energy gradient of 0.01 kcal/(mol Å) (ArgusLab 4.0.1, 2004).
Molecular docking: Structures of both protein and ligand molecule were extracted from the PDB data and were used in docking tests. After adding the hydrogen atoms, ligand molecules were minimized using the Universal Force Field (UFF) implemented in ArgusLab. For the docking tests, both ArgusDock and GADock were evaluated; then, the results were compared. Default setting of the scoring function and adjusted functions were used for the study. Furthermore, size of the binding site bounding box was determined automatically using ArgusLab (16.452x15.215x14.010 Å). Root-Mean-Square Deviation (RMSD) between the experimental and computational ligand structure were computed to evaluate the accuracy of the calculated poses; the calculated pose with RMSD that was less than or equal to 2.0 Å was defined as reasonable poses. The validated method of docking calculation then used to perform the docking of flavonoids and quercetin derivatives to iNOS binding site. Binding affinity was characterized by binding energy values (ΔG) and hydrogen bonds between ligands and the enzyme (Oda et al., 2007).
Predicting the affinities of quercetin derivatives to iNOS: The in vitro biological activity data reported as IC50 for inhibition of iNOS by the flavonoids were used for the current study (Olszanecki et al., 2002). The correlation curve of the binding energies (ΔG, kcal mol-1) of flavonoids with iNOS to the experimental activities (-log IC50) was constructed. The obtained regression equation then was used to calculate the predicted IC50 of quercetin derivatives (Zheng et al., 2006; Ji and Zhang, 2006).
Predicting the absorption and distribution properties using PreADMET: PreADMET program was accessed at http://preadmet.bmdrc.org/. Chemical structures of the compounds were drawn or uploaded from Molfile (*.mol). The program automatically calculated the predictive adsorption and distribution parameter we used, i.e., permeability for Caco-2 cell, HIA (human intestinal absorption) and plasma protein binding (PreADMET, 2010).
RESULTS
Accuracy of docking method: Arguslab has two docking engine types, i.e., ArgusDock and GADock. In addition, A-score is used as a scoring function. We compared the accuracy of ArgusDock and GADock, by measuring the Root Mean Square Deviation (RMSD) of the Cartesian coordinates of the atoms of the ligand (3-bromo-7-nitroindazole) in the docked and crystallographic conformations. A docking method is generally regarded as successful if the RMSD value is less than 2Å (Morris and Lim-Wilby, 2008). In present study, ArgusDock engine was failed to perform an accurate docking calculation, due to the RMSD value was 5.9067 Å. However, GADock engine gave a better result. Figure 2 shows the conformational superposition of 3-bromo-7-nitroindazole from the X-ray crystal structure of 3-bromo-7-nitroindazole–iNOS complex and that from the docking calculation by GADock engine. RMSD value between the two conformations is only 1.3494 Å, indicating that the parameter set for docking is capable of reproducing the X-ray structure. In addition, both of ligand (original ligand and that from the docking simulation) interact to the same residue of iNOS, i.e., Met368, so this engine then was used to perform the docking calculation of the flavonoids and quercetin derivatives.
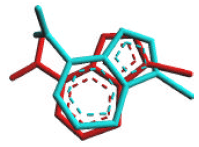 | |
| Fig. 2: | Conformational comparison of 3-bromo-7-nitroindazole from the crystal structure of 3-bromo-7-nitroindazole–iNOS complex (red) and that from the docking simulation using GADock engine (blue) |
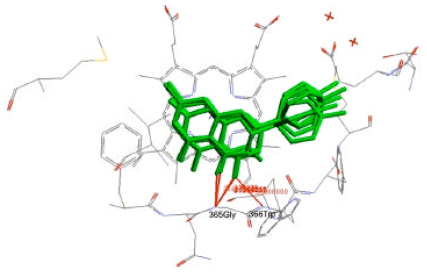 | |
| Fig. 3: | Superimposition of the binding conformations of flavonoids (green) on the binding site of iNOS. Hydrogen bonds are marked in red lines |
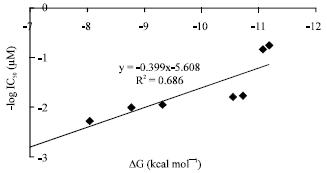 | |
| Fig. 4: | Correlation of predicted binding energy (ΔG, kcal mol-1) of flavonoids with iNOS to experimental activities (-log IC50) |
Molecular docking of flavonoids: Structures of selected flavonoids i.e., kaempferol, quercetin, apigenin, primuletin, catechin, hesperetin and naringenin (Table 1) were used as ligands for molecular docking to iNOS binding site. By using GADock method, the seven flavonoids were docked with iNOS and the binding affinity was characterized by binding energy (ΔG). Figure 3 shows the alignments of the binding conformations of the flavonoids to iNOS.
As shown in Fig. 4, there is a correlation between experimental values (expressed by IC50) and theoretical parameters (measured by binding energy), which justifies the present structural models and docking methods:
| (1) |
This correlation indicated that ΔG values calculated by GAdock method can be used to predict the iNOS inhibitory activities of quercetin derivatives.
| Table 2: | Structures quercetin derivatives, their binding energies and predicted inhibitory activities |
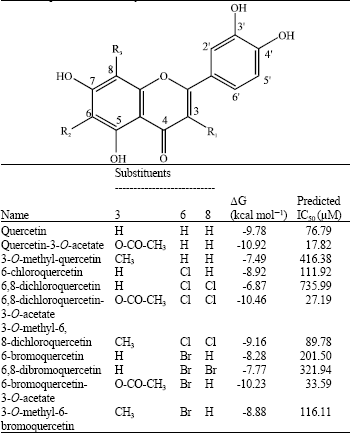 | |
Predicting the affinities of quercetin derivatives to iNOS: Applying the above equation, the predicted IC50 values of the quercetin derivatives (Table 2) were calculated. Figure 5 showed the interaction of 6, 8-dichloroquercetin-3-O-acetate to iNOS binding-site, compare to that of quercetin, binding energy of quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O- acetate were lower than that of quercetin, indicated that these compounds were proposed to be more potent as iNOS inhibitor.
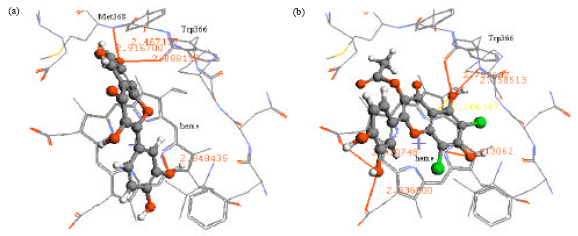 | |
| Fig. 5: | (a) Interaction of quercetin and (b) 6,8-dichloroquercetin-3-O-acetate to iNOS binding site |
| Table 3: | Predictive absorption and distribution properties of quercetin derivatives |
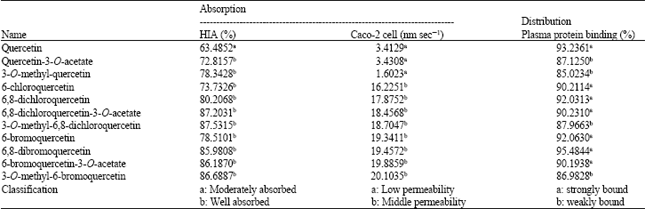 | |
Prediction of absorption and distribution properties: Table 3 presented the values of predictive absorption and distribution of quercetin and its derivatives. Quercetin was predicted as moderately absorbed and low permeable compound, as well as strongly bound. These were not good properties for oral drug candidate. On the contrary, predictive absorption and distribution properties of some quercetin derivatives which proposed more potential as iNOS inhibitor than quercetin, i.e. quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetat, generally were better than those of quercetin. Quercetin-3-O-acetate was predicted as well absorbed and weakly bound compound. In addition, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetat were predicted as well absorbed and middle permeability compounds.
DISCUSSION
Prediction of interaction between 3D structure of protein and different ligands by molecular docking helps researchers to find out important aspects such as the active sites, binding energies, etc. This in silico technique is very useful in the initial phase of drug discovery (Amir et al., 2010). ArgusLab is one of computational software equipped with two molecular docking tools, i.e. ArgusDock and GADock. Oda et al. (2007) reported that GADock is superior in terms of accuracy than ArgusDock. Our validation results based on the RMSD values also indicated that GADock gave better accuracy than ArgusDock.
After being validated, GADock engine was used to conduct the molecular docking between iNOS binding site and flavonoids. In general, the flavonoids consist of 2 benzene rings (A and B), which are connected by an oxygen containing pyrane ring (C). The X-ray structure of 3-bromo-7-nitroindazole, a known iNOS inhibitor, shows that the aromatic ring of 3-bromo-7-nitroindazole binds to the heme pocket of iNOS. In addition, the nitro moiety forms hydrogen bond with amide group of Met368. Flavonoids could be docked similarly as 3-bromo-7-nitroindazole with the planar region of A-ring of flavonoids oriented to the heme plane of iNOS. According to Rosenfeld et al. (2002), when bound to NOS, the planar system showed higher-affinity compared to the nonplanar system. The carbonyl-groups of the flavonoids form hydrogen bond with amide groups of Gly365 and Trp366.
Earlier study describing interaction of ligands to iNOS was reported by Francis et al. (2008). It was reported that the active site of NOS consists of four pockets, i.e. the substrate binding S pocket, the middle M pocket, the C1 pocket and C2 pocket in the substrate access channels. The residues Trp372 and Glu377 in the S pocket (iNOS) have been found to be the main residues with which the substrates (L-arginin) form hydrogen bond. Our results showed that the flavonoids form hydrogen bond with residues Gly365 and Trp366. However, there are clear structural differences between L-arginin and flavonoids as ligands and hence lead to different amino acid residues of iNOS taking part in the interaction.
Based on the study of interaction between flavonoids and iNOS binding site, the affinity of quercetin derivatives on iNOS binding site was predicted. It was obtained that energy binding values of quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetate were lower than that of quercetin, indicating that these compounds have higher affinity on iNOS binding site than quercetin. The iNOS inhibition properties of these compounds have not been reported. Most publications (Hu and Kitts, 2004; Hamalainen et al., 2007; Wan et al., 2009) reported inhibitory effects of flavonoids on iNOS genetic expression instead of iNOS inhibition properties of quercetin derivatives. The most relevant information with our work was reported by Chen et al. (2001) in which quercetin pentaacetate enhanced the iNOS activity to form Nitric Oxide (NO), compared to quercetin.
Absorption and distribution, the part of pharmacokinetics, were considered as important parameters to choose compounds as drug candidates. In this study, three parameters from PreADMET program were calculated for quercetin and a set quercetin derivatives. PreADMET featured prediction of absorption properties, including Caco-2 cell permeability as well as percent human intestinal absorption (%HIA). Caco-2 cell model is reliable in vitro models for the prediction of oral drug absorption, while HIA is the sum of bioavailability and absorption evaluated from ratio of excretion or cumulative excretion in urine, bile and feces. For distribution properties, we used the calculation of predictive plasma protein binding which available in PreADMET program. Only the unbound drug is available for diffusion or transport across cell membranes and also for interaction with a pharmacological target; therefore, plasma protein binding of a drug plays an important role in drug’s efficacy (Lee et al., 2003).
Although, quercetin has been demonstrated to perform some beneficial biological activities, the high-effective concentration and poor-absorptive properties limited its practical applications. Gugler et al. (1975) have studied the pharmacokinetic profile of quercetin. It was resulted that after i.v. administration, the protein binding of quercetin was higher than 98%. After oral administration no measurable plasma concentration of quercetin could be detected, nor was found in urine, either in unchanged or as metabolized form. It was proposed that the absorption of quercetin was very low. Our prediction value of pharmacokinetic properties of quercetin calculated by PreADMET showed the similar results. On the contrary, the pharmacokinetics profile of some quercetin derivatives, including quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetate, were predicted to be better that of quercetin. It was suggested that molecular modification by acetylation and halogenations increased the lipophilicity of quercetin and furthermore enhanced its oral absorption. In addition, these modifications also decrease the protein binding of quercetin.
CONCLUSIONS
Instead of ArgusDock, GAdock method was more suitable to predict the iNOS inhibitory activities of quercetin derivatives. By using GAdock method, computational docking of flavonoids to iNOS binding-site resulted to a correlation between experimental values (IC50) and theoretical parameters (binding energy). Quercetin-3-O-acetate, 6,8-dichloroquercetin-3-O-acetate and 6-bromoquercetin-3-O-acetat were proposed to be more potential as iNOS inhibitor than quercetin. The predictive absorption and distribution properties of these compounds i.e. human intestinal absorption, Caco-2 cell permeability and percent plasma protein binding were better than those of quercetin.
REFERENCES
- Amir, A., M.A. Siddiqui, N. Kapoor, A. Arya and H. Kumar, 2011. In silico molecular docking of influenza virus (PB2) protein to check the drug efficacy. Trends Bioinform., 4: 47-55.
CrossRefDirect Link - Chen, Y.C., S.C. Shen, W.R. Lee, W.C. Hou, L.L. Yang and T.J.F. Lee, 2001. Inhibition of nitric oxide synthase inhibitors and lipopolysaccharide induced inducible NOS and cyclooxygenase-2 gene expressions by rutin, quercetin and quercetin pentaacetate in RAW 264.7 macrophages. J. Cell. Biochem., 82: 537-548.
CrossRefPubMedDirect Link - Coskun, O., M. Kanter, F. Armutcu, K. Cetin, B. Kaybolmaz and O. Yazgan, 2004. Protective effects of quercetin, a flavonoid antioxidant, in absolute ethanol-induced acut gastric ulcer. Eur. J. Gen. Med., 1: 37-42.
Direct Link - Francis, S.M., A. Mittal, M. Sharma and P.V. Bharatam, 2008. Design of Benzene-1,2-diamines as selective inducible nitric oxide synthase inhibitors: A combined de novo design and docking analysis. J. Mol. Model., 14: 215-224.
CrossRef - Gugler, R., M. Leschik and H.J. Dengler, 1975. Disposition of quercetin in man after single oral and intravenous doses. Eur. J. Clin. Pharm., 9: 229-234.
CrossRef - Hamalainen, M., R. Nieminen, P. Vuorela, M. Heinonen and E. Moilanen, 2007. Anti-inflammatory effects of flavonoids: Genistein, kaempferol, quercetin, and daidzein inhibits STAT-1 and NF-ΚB activation, whereas flavone, isorhamnetin, naringenin, and pelargonidin inhihibit only NF-ΚB activation along with their inhibitory effect on iNOS expression and NO production in activated macrophages. Mediators Inflamm., 2007: 1-10.
CrossRef - Hu, C. and D.D. Kitts, 2004. Luteolin and luteolin-7-O-glucoside from dandelion flower suppress iNOS and COX-2 in RAW264.7 cells. Mol. Cell. Biochem., 265: 107-113.
CrossRef - Ji, H.F. and H.Y. Zhang, 2006. Theoretical evaluation of flavonoids as multipotent agents to combat Alzheimer's disease. J. Mol. Struct. THEOCHEM, 767: 3-9.
CrossRef - Knowles, R.G. and S. Moncada, 1994. Nitric oxide synthasein mammals. Biochem. J., 298: 249-258.
Direct Link - Korhonen, R., A. Lahti, H. Kankaanranta and E. Moilanen, 2005. Nitric oxide production and signaling in inflammation. Curr. Drug Targets Inflamm. Allergy, 4: 471-479.
PubMed - Morikawa, K., M. Nonaka, M. Narahara, I. Torii and K. Kawaguchi et al., 2003. Inhibitory effect of quercetin on carrageenan-induced inflammation in rats. Life Sci., 26: 709-721.
CrossRef - Oda, A., M. Okayasu, Y. Kamiyama, T. Yoshida, O. Takahashi and H. Matsuzaki, 2007. Evaluation of docking accuracy and investigastions of roles of parameters and each term in scoring functions for protein-ligand docking using arguslab software. Bull. Chem. Soc. Jap., 80: 1920-1925.
CrossRef - Olszanecki, R., A. Gebska, V.I. Kozlovski and R.J. Gryglewski, 2002. Flavonoids and nitric oxide synthase. J. Physiol. Pharmacol., 53: 571-584.
Direct Link - Peng, Y., Z. Deng and C. Whang, 2008. Preparation and prodrug studies of quercetin pentabenzensulfonate. Yakugaku Zasshi, 128: 1845-1849.
Direct Link - Rosenfeld, R.J., E.D. Garcin, K. Panda, G. Andersson and A. Aberg et al., 2002. Conformational changes in nitric oxide synthases induced by chlorzoxazone and nitroindazoles: Crystallographic and computational analyses of inhibitor potency. Biochemistry, 41: 13915-13925.
PubMed - Wan, L.L., J. Xia, D. Ye, J. Liu, J. Chen and G. Wang, 2009. Effects of quercetin on gene and protein expression of NOX and NOS after myocardial ischemia and reperfusion in rabbit. Cardiovasc. Ther., 27: 28-33.
PubMed - Zheng, M., Z. Zhang, W. Zhu, H. Liu, X. Luo, K. Chen and H. Jiang, 2006. Essential structural profile of a dual functional inhibitor against cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX): Molecular docking and 3D-QSAR analyses on DHDMBF analogues. Bioorganic Med. Chem., 14: 3428-3437.
CrossRef