
Sarita Agrawal
Rugnta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Tapan Kumar Giri
Rugnta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Dulal Krishna Tripathi
Rugnta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Ajazuddin
Rugnta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
Amit Alexander
Rugnta College of Pharmaceutical Sciences and Research, Kohka-Kurud Road, Bhilai, Chhattisgarh, India
LiveDNA: 91.5283
ORCID: 0000-0003-0391-7650
American Journal of Drug Discovery and Development
Year: 2012 | Volume: 2 | Issue: 4 | Page No.: 143-183







ABSTRACT
Self micro-emulsifying drug delivery systems (SMEDDS) are vital tool for enhancement of oral bioavailability of hydrophobic drugs. These systems are currently of interest to the researchers because of their significant capability to act as drug delivery vehicles by incorporating a extensive range of drug molecules. The present communication embodies approaches in the design of lipid based formulation, evaluation processes, mechanism involved there in, updated with latest findings from literature reports and patents. Also, this comprehensive review offers an explicit discussion on vital possibilities of the SMEDDS in bioavailability improvement of various drugs. A pseudo ternary phase diagram is used for identifying the micro-emulsification region. Thus, this current article provides an updated compilation of extensive information and result on all the unexplored areas of the self micro emulsifying drug delivery systems, thus encouraging the researchers to accelerate their research work in this direction for the development and enhancement of dissolution profile of hydrophobic drugs and pay a novel approach to pharmaceutical research.
PDF Abstract XML References Citation
Received: January 23, 2012;
Accepted: March 14, 2012;
Published: May 16, 2012
How to cite this article
Sarita Agrawal, Tapan Kumar Giri, Dulal Krishna Tripathi, Ajazuddin and Amit Alexander, 2012. A Review on Novel Therapeutic Strategies for the Enhancement of Solubility for Hydrophobic Drugs through Lipid and Surfactant Based Self Micro Emulsifying Drug Delivery System: A Novel Approach. American Journal of Drug Discovery and Development, 2: 143-183.
DOI: 10.3923/ajdd.2012.143.183
URL: https://scialert.net/abstract/?doi=ajdd.2012.143.183
DOI: 10.3923/ajdd.2012.143.183
URL: https://scialert.net/abstract/?doi=ajdd.2012.143.183
INTRODUCTION
Oral route is the easiest, most convenient route for non invasive administration and the major route of drug delivery for the chronic treatment of many diseases (Reddy and Murthy, 2002). In current years, new chemical entities exhibit poor aqueous solubility which in turn leads to low oral bioavailability (Robinson, 1996). Formulation of poorly aqueous soluble drugs is a challenging job to the pharmaceutical scientists as result of modern drug discovery technique and oral delivery of such drugs is frequently associated with low bioavailability, high inter subject variability and lack of dose proportionality (Dey et al., 2009; Kanika et al., 2010; Giri et al., 2010b). The formulation technique plays an important role in overcoming this shortcoming of poorly water soluble drugs, to encountered these problems, various formulation strategies are reported including use of surfactants, pulverization, crystal polymorphism selection, salt formation, solid dispersion, mixed pulverization, complex formation agent like cyclodextrin, emulsion, micro emulsion, liposome, particle size, nanoparticles, micro and nano spheres, lipids carriers, use of prodrug, drug derivatization, solution phase studies and permeation enhancers to improve the dissolution rate of the drug (Mandal and Mandal, 2011; Badawi et al., 2011; Seedher and Sharma, 2007; Wanwimolruk et al., 1992; Amarji et al., 2007). In recent years, a lot interest has given on lipid-based formulations to enhance the oral bioavailability of poorly aqueous soluble drug compounds (Burcham et al., 1997; Patel et al., 2011). Lipid formulations for oral administration of drugs generally consist of a drug dissolved in oils, partial glycerides, surfactants or co-surfactants. The principal mechanism of action which leads to improved bioavailability is usually avoid the slow dissolution process which limits the bioavailability of hydrophobic drugs from solid dosage forms (Pouton, 2000; Tang et al., 2007). The Self-Dispersing Lipid Formulations (SDLFs) is one of approaches to overcome the formulation difficulties of various hydrophobic drugs and to improve the oral bioavailability of poorly absorbed drugs. The SDLFs are mainly two type’s i.e., Self Emulsifying Drug Delivery System (SEDDS) and Self Micro Emulsifying Drug Delivery System (SMEDDS) (Gershanik and Benita, 2000). SMEDDS are isotropic and thermodynamically transparent stable solutions consisting of an oil, surfactant, co-surfactant and drug mixtures which form oil-in-water microemulsions when mixed with aqueous phase under mild stirring. Potential advantages of these systems include not only enhanced drug solubilization but also improved release and absorption properties due to the already dissolved form of the drug in the formulation and the resulting small droplets size, providing a large interfacial surface area for drug absorption (Farah et al., 1994; Craig et al., 1995). To the improved dissolution of drugs by SMEDDS, one more factor contributing to the increasing bioavailability is that lymphatic transport is responsible for a portion of the complete drug uptake as well (Porter et al., 2007). Figure 1 illustrates the oral drug absorption of self-emulsifying formulations from the GI mucosa to systemic circulation. It can also be changed into granules, pellets, powders for dry filled capsules or tablet preparations and also include into Ca-alginate microcapsules (Nazzal and Khan, 2006; Abdalla et al., 2008; Serratoni et al., 2007; Tan et al., 2009).
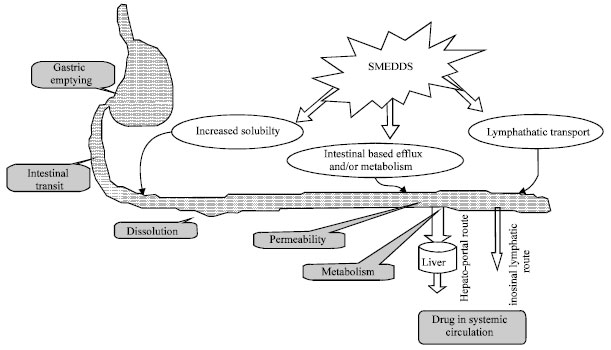 |
| Fig. 1: | Self-emulsifying formulations enhancing the bioavailability of drugs through oral absorption |
MICROEMULSION
The concept of microemulsion was first introduced by Hoar and Schulman in 1943 (Hoar and Schulman, 1943). Microemulsion is a system of water, oil and amphiphilic compounds (surfactant and co-surfactant) which is a transparent, optically isotropic and thermodynamically stable liquid (Lawrence and Rees, 2000). In practice ‘micro emulsions’ are frequently identified by equilibrium phase studies as systems which are optically transparent to the eye, however, contain a extensive mass of both oil and water (Pouton, 1997). Micro emulsions were prepared by mixing a suitable quantity of aqueous solution with the organic phase containing the surfactant solution (Yadav et al., 2008). They help in the improvement of drug bioavailability, protection against enzymatic hydrolysis and decrease toxicity. The only problem with microemulsion is poor palatability and moreover due to their water content, microemulsions cannot be encapsulated in soft and hard gelatin (Shinde et al., 2011). Hence, there is a need for delivery of hydrophobic drug is Self-Micro emulsifying Drug Delivery System (SMEDDS).
SMEDDS
Self Micro Emulsifying Drug Delivery System (SMEDDS) are defined as isotropic mixtures of oils, surfactants, along with co-solvents/surfactants that have a unique ability of forming fine oil-in-water (o/w) micro emulsions upon moderate mixing of these ingredients in aqueous media, such as GI (Gastrointestinal) fluids (Patravale et al., 2003). Self emulsifying drug delivery systems (SEDDS) SEDDS produce not clear emulsions with a droplet size between 100 and 300 nm while SMEDDS form clear micro emulsions with a droplet size of less than 50 nm also the concentration of oil in SMEDDS is less than 20% as compared to 40-80% in SEDDS. According to the studies of Self-Emulsifying Drug Delivery Systems (SEDDS) formed using surfactants of HLB<12 and Self-Micro Emulsifying Drug Delivery Systems (SMEDDS) formed with surfactants of HLB>12. Thus, for lipophilic drug compounds that exhibit dissolution rate-limited absorption, these systems may offer an improvement in the rate and extent of absorption and result in more reproducible blood-time profiles (Gursoy and Benita, 2004; Pouton, 2000). The key step is to find a suitable oil surfactant mixture that can dissolve the drug within the required therapeutic concentration. The SMEDDS mixture can be filled in either soft or hard gelatin capsules. A typical SMEDDS formulation contains oils, surfactants and if required an antioxidants. Often co-surfactants and co-solvents are added to improve the formulation characteristics (Khoo et al., 1998; Aqil et al., 2011).
Benefits of SMEDDS:
| • | Enhancement in oral bioavailability e.g., ketoprofen |
| • | Reduction in inter-subject and intra-subject variability and food effects e.g., cyclosporine |
| • | SMEDDS has capability to deliver peptides that are prone to enzymatic hydrolysis in GIT |
| • | For both liquid and solid dosage forms. e.g., Progesterone |
| • | Ease of manufacture and scale- up (Patel and Sawant, 2009) |
Limitations of SMEDDS:
| • | Chemical instabilities of drugs and high surfactant concentrations |
| • | The large amount of surfactant in self-emulsifying formulations (30-60%) irritates GIT |
| • | Moreover, volatile co solvents in the conventional self-emulsifying formulations are known to migrate into the shells of soft or hard gelatin capsules, resulting in the precipitation of the lipophilic drug |
MECHANISM OF SELF EMULSIFICATION
The exact mechanism of self-emulsification is not yet well explained. When the energy required for increasing the surface area of dispersion is less than the change in entropy required for dispersion, self emulsion takes place. Moreover, the free energy of a traditional emulsion formation and the energy required for increasing surface area are directly related as shown below:
where, ΔG is the free energy associated with the process (ignoring the free energy of mixing), N is the number of droplets having radius, r and s is the interfacial energy. The above equation shows that spontaneous formation of interface between oil and aqueous phase is thermodynamically stable (Reiss, 1975). Gershanik and Benita (2000) explained the spontaneous formation of emulsion, i.e., self-emulsification, in terms of the free energy required to form the emulsion which is either very low and positive, or negative.
The ease of emulsification has been quantitatively measured by Mustafa and Groves. The turbidity of the oil-surfactant system in a water stream was monitored by using Phosphated nonyl phenoloxylate (PNE) and Phosphated Fatty alcohol Ethoxylate (PFE) in n-hexane. They proposed the relation between emulsification process and (i) how easily water can penetrates into the oil-water interface (ii) formation of liquid crystalline phase that results swelling at the interface.
Pouton has proposed a relationship between the emulsification properties of the surfactant and phase inversion behavior of the system. For example, the temperature of the oil in water system, stabilized by using non-ionic surfactant(s) is increased; the cloud point of the surfactant would be attained followed by phase inversion. The surfactant is highly mobile at the phase inversion temperature; hence, the o/w interfacial energy is minimized, leading to a reduction in energy required to bring about emulsification.
CLASSIFICATION OF LIPID FORMULATION
The main purpose of the lipid formulation classification system is to enable in vivo studies to be interpreted more readily and, subsequently, to facilitate the identification of the most appropriate formulations for specific drugs with reference to their physicochemical properties (Pouton and Porter, 2008). Each lipid formulation type has specific features as described in Table 1 by Pouton (2006).
| Table 1: | Characteristics, advantages and disadvantages of lipid formulations |
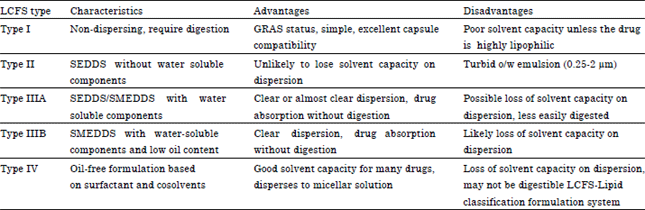 | |
| Source: Pouton (2006) | |
Types of lipid formulations: Type I formulations consist of formulations solubilized drug in triglycerides and/or mixed glycerides or in an oil-in-water emulsion stabilized by little concentration of emulsifiers such as 1% (w/v) polysorbate 60 and 1.2% (w/v) lecithin. Generally, these systems show poor initial aqueous dispersion and require digestion by pancreatic lipase/co-lipase in the GIT to produce more amphiphilic lipid digestion products and promote drug transfer into the colloidal aqueous phase. Type 1 formulations therefore are a good option for drugs having sufficient solubility in oils. Valproic acid has been formulated in soft gelatin capsule containing corn oil as lipidic component.
Type II formulations are referred to as SEDDS. SEDDS are isotropic mixtures of lipids, surfactants (HLB<12), co-surfactant and the drug which form oil-in water emulsions under gentle agitation subsequent dilution with aqueous phases. Self-emulsification is usually obtained at surfactants contents above 25% (w/w). However, at higher surfactants concentration (greater than 50-60% (w/w)), the progress of emulsification may be hindered by the formation of viscous crystalline, gels at the oil/water interface. A Type II system has received limited attention and no marketed products have emerged. One reason may be that the most effective surfactants for Type II formulation do not appear on the FDA list of inactive ingredients.
Type III formulations are generally referred as self microemulsifying drug delivery systems (SMEDDS). It consist of oils, hydrophilic surfactants (HLB>12) and co-solvents. Type III formulations are further divided into Type IIIA and Type IIIB formulations. Later comprises of higher amount of hydrophilic surfactants and co-solvents and lesser lipid content, as compared to Type IIIA. Type IIIB formulations cause greater risk of drug precipitation on dispersions given their high content of hydrophilic surfactants and co-solvents. An example of marketed Type III formulation is Neoral® (Novartis) cyclosporine formulation. This formulation comprises of corn oil glycerides, cremophor RH40, glycerol, propylene glycol and ethanol.
Type IV systems are basically pure surfactants or mixtures of surfactants and co-solvents. Formulation of poorly water-soluble drugs in pure co-solvents is likely to result in precipitation of the drug. An example of a commercial Type IV formulation is Agenerase® (GlaxoSmithKline), a capsule formulation of the HIV protease inhibitor amprenavir containing tocopherol polyethylene glycosuccinate (TPGS) as a surfactant and PEG 400 and propylene glycol as co-solvents (Porter et al., 2008; Carrigan and Bates, 1973; Myers and Stella, 1992; Patel et al., 2011; Wanwimolruk and Levy, 1987; Arif et al., 1996). Table 2 provides a list of various lipid formulations in commercial circulation available.
SELECTION OF COMPONENTS FOR SMEDDS
The crucial challenges to any oral formulation design program is maintaining drug solubility within the gastrointestinal tract and, in particular, maximizing drug solubility within the primary absorptive site of the gut (O’Driscoll and Griffin, 2008). Lipid based formulations offer a potential platform for improving oral bioavailability of drugs especially those belonging to biopharmaceutical Classification System (BCS) class II and class IV. Class II drugs are poorly water soluble drugs with high permeability but once they are dissolved; they absorbed over the gastro- intestinal membrane, and Class IV compounds are poorly soluble with poor permeability, respectively (Tapas et al., 2011). The basic criteria for selection of components of lipid formulation are; the lipophilicity of the drug, with solubility in pharmaceutically-acceptable lipid excipients which should be sufficient to allow the entire dose of the drug to be administered in a single dosage unit. Another reason for achieving success of a lipid based formulation by use of a strong positive food effect.
| Table 2: | List of marketed product of lipid formulation |
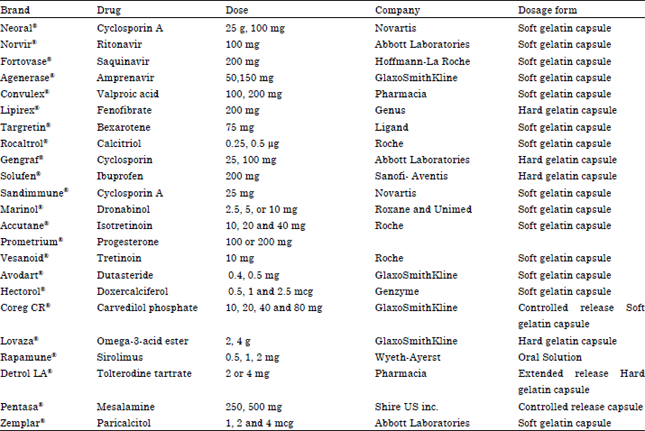 | |
| Source: Chakraborty et al. (2009) | |
| Table 3: | Application of SMEDDS in various BCS category drugs |
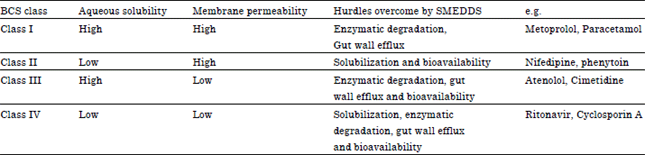 | |
| Source: Kanika et al. (2010) | |
The presence of fatty meal in stomach instead of empty stomach favors the absorption of drug from the lipid based formulation because the absorption of lipophilic drug usually exhibit dissolution-rate-limited. SMEDDS can improve the rate and extent of absorption resulting in reproducible blood time profiles (Hauss, 2007). These systems can help in solving the under-mentioned problems of all the categories of BCS class drugs, as depicted in Table 3. To explain the trends for oral absorption Lipinski’s rule of five has been widely used as a qualitative predictive model. The rule of five explains poor absorption or poor permeation in terms of situation where there are more than five H-bond donors, there are more than ten H-bond acceptors, the molecular weight >500 and the calculated log p >5. Both BCS and Lipinski’s rule of five are useful, mainly at the primary screening stage but they have limitations. It is considered that the rule of five only applicable to compounds which are not substrates for active transporters, and with increasing evidence suggesting that most drugs are substrates for some efflux or uptake transporters, this limitation might be notable (Kohli et al., 2010). For design a lipidic systems Log P can be consider as the prime characteristics. For lipidic systems higher log P (more than 4) values are desirous. For e.g., cinnarizine, a lipophilic drug, having log P values greater than 5 is strong candidate for SMEDDS. Among physicochemical characteristics melting point and dose play a major role. Low melting point and low dose are desirable for development of lipidic systems. Drugs high melting point having with low log P values (around 2) is not suitable for SMEDDS.
EXCIPIENTS USED IN SMEDDS
As described, a SMEDDS pre-concentrate can contain four categories of components: drug, lipids, surfactants and co solvents. Commonly used lipids, surfactants and co-solvents are listed in Table 4.
Oil: The oil represents one of the most essential excipients in the SMEDDS formulation not only because it can solubilize the necessary dose of the lipophilic drug or assist self emulsification but also and mainly because it can improve the fraction of lipophilic drug transported via the intestinal lymphatic system, thereby increasing absorption from the GI tract depending on the molecular nature of the triglyceride (Kimura et al., 1994). As a result, triglycerides such as medium chain and long chain with different degree of saturation have been used for the solvation of hydrophobic therapeutic agent in the design of SMEDDS (Constantinides, 1995). Long-chain triglycerides are derived from vegetable sources such as soybean or safflower oil, whereas MCTs are obtained by the re-esterification of fractionated coconut oil fatty acids with glycerin (Angare et al., 2012). Semisynthetic derivatives form good emulsification systems when used with a large amount of solubility enhancing surfactants approved for oral administration (Gershanik and Benita, 2000; Devani et al., 2004). Patravale et al. (2003) demonstrated that because of high fluidity, improved solubilizing potential and self-microemulsification potential these excipients form good emulsification systems. The stability of emulsion also depends on the rheology and characteristics of the oil (Anisa et al., 2010). Vegetables oil like Olive oil, Peanut oil, Safflower oil, Sesame oil, Soybean oil, Wheat germ oil, rice bran oil etc. (Pogori et al., 2008).
Surfactants: A surfactant is an amphiphilic agent formed by two parts with different affinities for the solvents. One of them has affinity for water (polar solvents) and the other has for oil (non-polar solvents) widely used for industrial, agricultural, food, cosmetics and pharmaceutical application such as emulsifying, solubilizing agent and enhancer (Gharaei-Fathabad, 2011; Noudeh et al., 2008). Surfactants used to stabilize microemulsion system may be: (i) non-ionic, (ii) zwitterionic, (iii) cationic, or (iv) anionic surfactants. Combinations of these, particularly ionic and non-ionic, can be very effective at increasing the degree of the microemulsion region (De Gennes and Taupin, 1982; Ajazuddinm, 2010). Surfactant also play vital role in the structure of colloidal sized cluster in solution, known as Micelles (Gokturk and Var, 2011). Anionic and nonionic surfactant mixtures are responsible for possible synergism (combined effect) in Critical Micelle Concentration (CMC) thus; Synergism of both surfactants was sought in presence of oil phase (Muherei and Junin, 2009; Tripathi et al., 1994). Non-ionic surfactants are identified to be less toxic compared to ionic surfactants. Various non-ionic surfactants such as the polysorbates like Tween 40, 60, 80 and polyoxyls which cover the HLB range from 2 to 18, may be used in combination with lipid excipients to facilitate self-emulsification or micro-emulsification (Hauss, 2007; Chen et al., 2011).
| Table 4: | List of excipients |
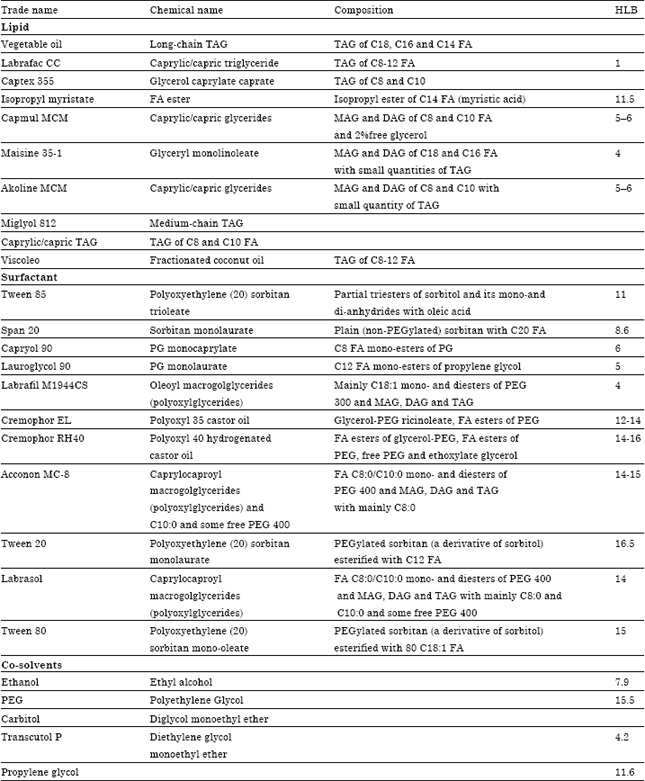 | |
| Source: Mullertz et al. (2010) | |
The surfactant used to enhance the bioavailability by various mechanisms including: improved drug dissolution in the gastrointestinal fluids, especially in the presence of bile salts, lecithin and lipid digestion mixtures, increased intestinal epithelial permeability, increased tight junction permeability and decreased/inhibited p-glycoprotein drug efflux (Patel and Sawant, 2009; Giri et al., 2010a). Emulsifier, a subset of surfactants which improves in machinability, strengthening and shelf life extension (Hoque et al., 2009). This can avoid precipitation of the drug within the GI lumen and for prolonged existence of drug molecules. In self-emulsifying formulations the usual surfactant concentration ranged from 30 to 60% w/w of the formulation. A large quantity of surfactant may irritate the GI tract (Tang et al., 2007).
Co-surfactant: In SMEDDS, usually co-surfactant of HLB value 10-14 is used with surfactants together to diminish the oil water interface, fluidize the hydrocarbon region of the interfacial film and allow the spontaneous formation of micro emulsion. The choice of co-surfactant and surfactant is critical not only to form the formation of microemulsion but also to solubilization in microemulsions (Patravale et al., 2003; Ozawa et al., 1986).
Co-solvent: The role of co-solvents in lipid based formulations mainly in SMEDDS is to assist the dispersion process and in earlier dispersion rates (Gursoy and Benita, 2004). Gershanik and Benita (2000) mentioned in their review about alcohol and other volatile co-solvent free self emulsifying micro emulsion formulations are known to migrate into the shells of soft gelatin, or hard, sealed gelatin capsules, resulting in the precipitation of the lipophilic drug.
Consistency builder: Tragacanth, cetyl alcohol, stearic acid or beeswax can be added to modify the stability of the emulsion (Osol, 1975).
Polymer: Inert polymer matrix representing from 5 to 40% of composition relative to the weight, which is not ionizable at physiological pH and being capable of forming matrix are used for the formulation of sustained release SMEDDS. Examples are hydroxypropylmethyl cellulose and ethyl cellulose (Barthelemy and Benameur, 2001; Alexander et al., 2011).
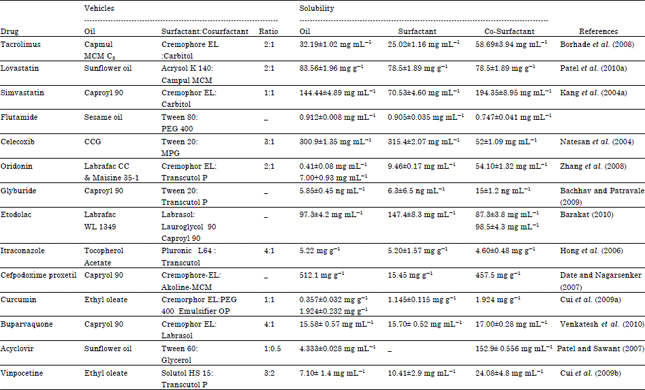
Table 5 shows list of various drug with their respective solubility in different vehicles like oil, surfactant and co-surfactant.
PSEUDO-TERNARY PHASE DIAGRAM STUDY
Pseudo-ternary phase diagrams are useful tools to determine the composition of an aqueous phase, oil phase and surfactant: co-surfactant phase that will yield a Micro Emulsion (ME), Liquid Crystal (LC) and coarse emulsion (EM). Each corner of diagram typically represents 100% of the particular component. It is constructed to define the extent and nature of micro emulsion region. The different phases are mix in different proportion to constructed the phase diagram and identify micro emulsion region. Since, four chemical species were incorporated in micro emulsion, one of the components (co-surfactant) is in fixed ratio with surfactant. Each of the three components for a system is titrated with the aqueous phase until a phase changes between micro emulsion and two phases of mixture was observed. Further addition of water it form the LC were detected under gentle stirring. By continuing the addition of water LC disappeared. However, unlike the first situation the mixture was somewhat cloudy and opaque which form the coarse emulsion (Li et al., 2005; Zadeh et al., 2010). An optimized formula for finding out region of microemulsion with the help of titration shows in Table 6.
| Table 5: | Drugs with their respective solubility in various vehicles |
 | |
| Table 6: | Titration chart to find out microemulsion region |
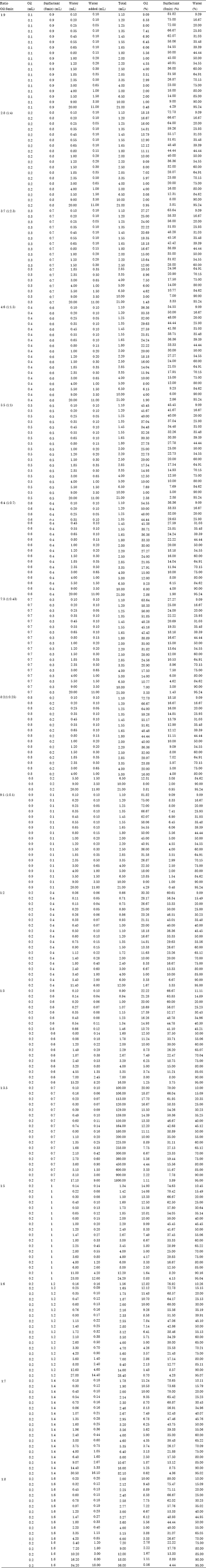 | |
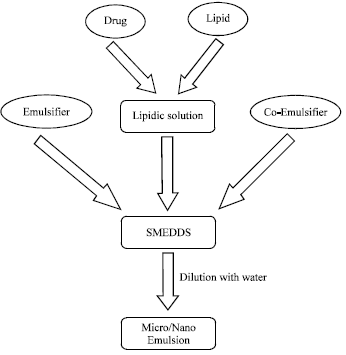 |
| Fig. 2: | The general strategy of formulating self micro-emulsifying systems and their subsequent conversion to micro/nano emulsions |
After optimization of microemulsion region we can draw the phase diagram with the help of software like Tri-plot 4.1.2, Chemix. For down loading the software please refer the given link home.c2i.net/astandne/help-htm/dwnload1.htm,mypage. iu.edu/~tthomps/programs/.
GENERAL METHOD FOR PREPARATION OF SMEDDS
Figure 2 illustrates the usual methodology pathways to prepare SMEDDS formulations and the formation of the micro-/nanoemulsions following their dilution.
| • | The solubility of the drug in different oil, surfactants and co solvents |
| • | The selection of oil, surfactant and co solvent based on the solubility of the drug |
| • | Preparation of the phase diagram |
| • | The formulations were prepared by initially mixing oil with surfactant at 50-60°C. Drug compounds were then dissolved into the mixture of surfactant and oil by constant stirring and kept at 50°C until a clear solution was obtained. All mixtures stayed clear at room temperature (Thia et al., 2009) |
| Table 7: | Literature Updates on various reports of Type II & III LFCS designed for the oral delivery of lipophilic drugs |
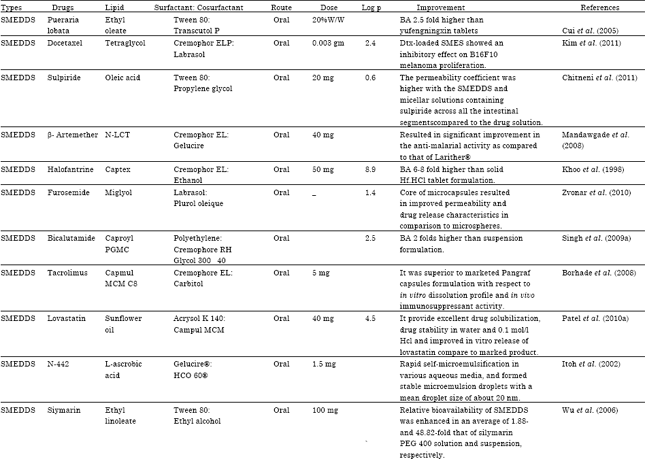 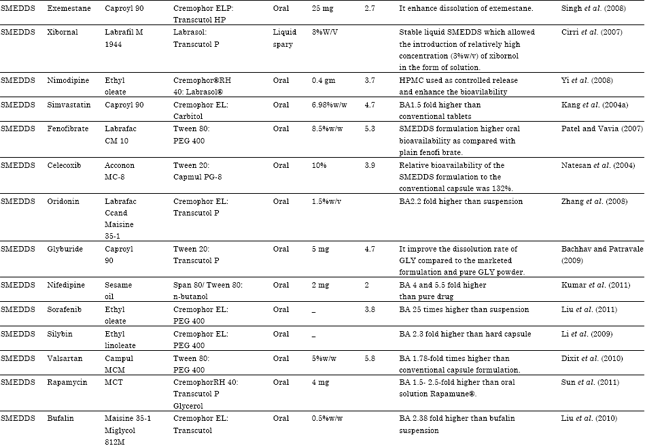 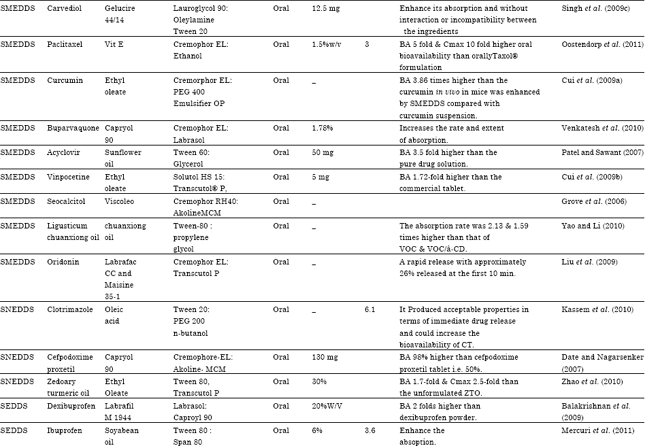 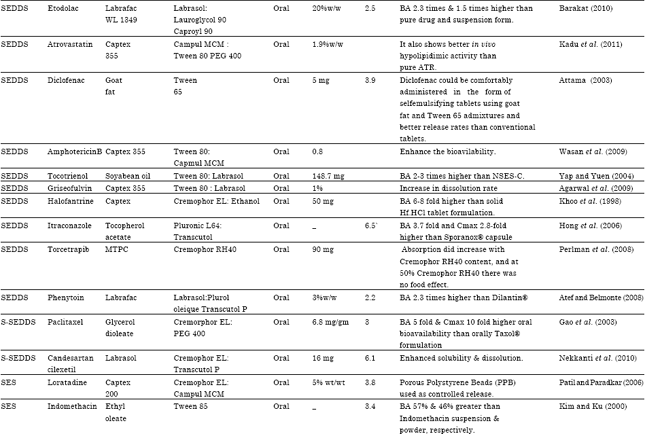  | |
| SMEDDS: Self micro emulsifying drug delivery system, S-SMEDDS: Solid self micro emulsifying drug delivery system, SEDDS: Self emulsifying drug delivery system, SNEDDS: Self nano emulsifying drug delivery system, SES: Self emulsifying system, SEF: Self emulsifying formulation, S-SEDDS: Solid self emulsifying drug delivery system, N-LCT: Natural lipophile, HCO: Hydrogenated castor oil, PEG: Polyethylene glycol, NSAID: Non steroidal anti inflammatory drug, MTPC: MCT/triacetin/polysorbate 80/capmulMCM, MCT: Medium chain triglyceride, PPB: Porous polystyrene beads, GLY: Glyburide, BA: Bioavilability, Dtx: Docetaxel | |
A Literature Updates on various reports of Type II and III LFCS designed for the oral delivery of lipophilic drugs for bioavailability enhancement as shown in Table 7.
FACTOR AFFECTING SMEDDS
Drug dose: Usually drugs having high dose are not preferred for developing SMEDDS. However, such drug if extremely soluble in any components of SMEDDS particularly in lipid phase. The drug which are not well soluble both in water and oil, and also posses low Log P value (around 2) are not suitable candidates for SMEDDS.
Drug solubility in oil phase: Solubility of the drug in oil phase greatly influenced the ability of SMEDDS in maintaining the drug in solution state. When the drug is solubilized by the use of surfactant and co surfactant the dilution of SMEDDS can lead to lowering the solvent capacity of surfactant or co surfactant, their by resulting precipitation.
Equilibrium solubility: For assessment of possibilities of precipitation in the gut equilibrium solubility measurement can be employed. Poutons study reveals that such formulation can take up to 5 days to reach equilibrium and that the drug can remain in a super saturated state up to 24 h after the initial emulsification event (Patel et al., 2010b).
Polarity of lipid phase: The polarity of lipid phase is one of the factors influencing the release of drug from the microemulsion. HLB, chain length and degree of unsaturation of fatty acid, molecular weight of the lipophilic portion and concentration of the emulsifier are factors for the polarity of droplets. The polarity indicates the affinity of the drug towards solvent, oil or water and the type of forces involved. The high polarity will promote rapid rate of release of the drug into the aqueous phase. Sang-Cheol et al. observed that the rate of release of Idebenone from SMEDDS is dependent upon the polarity of oil phase used. The highest release was obtained with the formulation that had oily phase with highest polarity (Kim et al., 2000).
Charge of emulsion droplets: Multiple physiological studies have shown the apical potentials of absorptive cells, ands of other cells in the body, are negatively charged compared to the mucosal solution in the lumen. Gershanik and Benita have shown that positively charged emulsion droplets formed by adding oleylamine (OA) to appropriate SEDDS undergo electrostatic interaction with the Caco-2 monolayer and the mucosal surface of the everted rat intestine (Gershanik et al., 2000). This formulation enhanced the oral bioavailability of progesterone in young rats. Benzoic acid had a dual function on the SEDDS; it could improve the self-emulsifying performance of Self-Emulsifying Oily Formulations (SEOFs) and Self-Microemulsifying Oily Formulations (SMEOFs) in 0.1 N HCl due to formation of a positively charged emulsion (Gershanik and Benita, 1996).
BIOPHARMACEUTICAL ASPECTS
Comprehensive literature survey reveals that certain lipids alone or with food can increase the bioavailability of some drugs. With incompleteness few explanations in support have been placed:
Alterations (reduction) in gastric transit: Increase in gastric resistance time shows the delivery of the drug of it site of absorption. There by the time for dissolution visa vis absorption is increased.
Increases in effective luminal drug solubility: The presence of lipids in the GI tract stimulates an increase in the secretion of Bile Salts (BS) and endogenous biliary lipids including phospholipids (PL) and Cholesterol (CH), leading to the formation of BS/PL/CH intestinal mixed micelles and an increase in the solubilisation capacity of the GI tract. However, intercalation of administered (exogenous) lipids into these BS structures either directly (if sufficiently polar), or secondary to digestion, leads to swelling of the micellar structures and a further increase in solubilisation capacity (Porter and Charman, 2001a).
Stimulation of intestinal lymphatic transport: For highly lipophilic drugs, lipids may enhance the extent of lymphatic transport and increase bioavailability directly or indirectly via a reduction in first-pass metabolism (Porter and Charman, 2001b).
Changes in the biochemical barrier function of the GI tract: It is evident some lipids and surfactants have ability to minimize the activity of intestinal efflux transporters, as sign of p-glycoprotein efflux pump. Similarly, the extent of enterocyte-based metabolism diminished (Benet and Cummins, 2001; Dintaman and Silverman, 1999; Nerurkar et al., 1996).
Changes in the physical barrier function of the GI tract: Although the passive intestinal permeability does not affect the bioavailability of majority of lipophilic, poorly soluble drugs, their permeability may be increased by certain mixtures of lipids, lipid digestion products and surfactants (Aungst, 2000; Muranishi, 1990).
Effect of oils on the absorption: Such formulations form a fine oil-in-water emulsion with gentle agitation which may be provided by gastrointestinal motility. A SES also improves the reproducibility of the plasma level-time profile. The effect of lipids on the bioavailability of orally administered drugs is highly complex due to numerous mechanisms by which the lipids can alter the biopharmaceutical characteristics of the drug. They include a decreased rate of gastric emptying, an increased dissolution rate of the drug and solubility in the intestinal fluid and the formation of lipoproteins promoting the lymphatic transport of highly lipophilic drugs (Craig, 1993; Hauss et al., 1998; 2007).
MECHANISM OF BIOAVAILABILITY ENHANCEMENT FROM SMEDDS
Most SMEDDs are based on triglycerides; it is helpful to consider the mechanisms by which SMEDDs are absorbed from the GI tract (Fig. 3a, b).
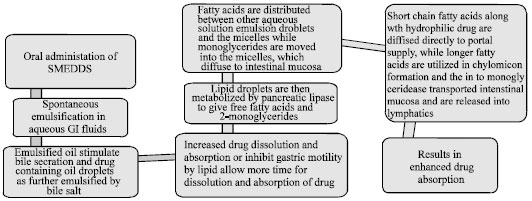
The absorption of fats from the GIT: Constantinides (1995) demonstrated that triglyceride molecules are fatty acid esters of glycerol. The ester groups of the triglycerides are prone to hydrolysis and this represents the major initial route of metabolism within the GI tact. On ingestion of the triglycerides, the lipids enter the stomach. Some hydrolysis may occur in the stomach due to the presence of gastric lipase. On entering the upper section of the small intestine, two processes occur. The fat droplets are further emulsified by the bile salts, monoglycerides, cholesterol, lecithin and lysolecithin to produce droplets with a diameter of approximately 0.5-1 μm. The triglyceride droplets are then metabolized by pancreatic lipase, to free fatty acids and 2-monoglycerides, the last two contributing to the digestion process as they themselves are emulsifying agents.
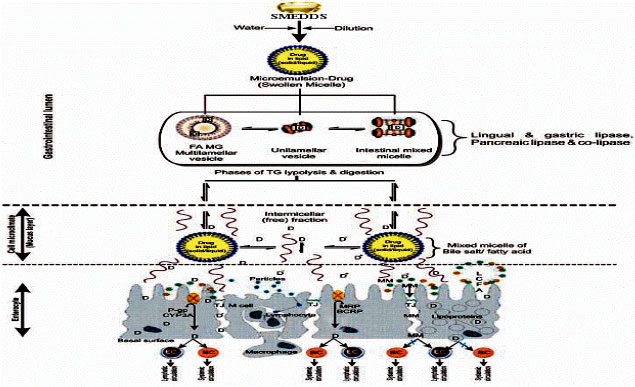 |
| Fig. 3a: | Diagrammatic representation mechanistic pathways for transportation of drugs across the GI lumen using SMEDDS |
 |
| Fig. 3b: | Mechanisms proposed for bioavailability enhancement of drugs |
The fatty acids are distributed between the aqueous solution, the emulsion droplet and the micelles, while the monoglycerides are incorporated into the micelles and are believed to swell the structure, allowing incorporation of other water insoluble components. The micelles then diffuse through the gut contents to the intestinal mucosa. Once in the intestinal mucosa, the monoglycerides are resynthesized into triglycerides and covered with a layer of lipoprotein, cholesterol and phospholipids. The resulting particles are released into lymphatic system. Short chain fatty acids may diffuse directly into the portal supply.
Bioavailability of drugs from oily vehicles: According to studies of (Benet and Cummins, 2001) compared the absorption of griseofulvin from commercial tablets, a corn oil emulsion (equivalent to 12 g oil) and an aqueous suspension in humans. The authors found that emulsion gave a much more rapid excretion of griseofulvin metabolite, desmethylgriseofulvin. The authors suggested that factors such as the inhibition of gastric motility caused by the presence of the lipid might have allowed more time for dissolution and absorption of drug. Alternatively, the presence of the emulsified oil may have stimulated bile secretion, which may have improved bioavailability. Later hypothesis have included increased mucosal permeability via incorporation of lipids from mixed micelles and enhanced mesenteric lymph flow.
Drug absorption from SMEDDs: The authors suggested that as the oil phase was a medium chain triglyceride, lymphatic uptake was unlikely to be enhanced; hence, the drug absorption may be a function of the increased surface area for dissolution provided by the emulsion. The authors also suggested that the presence of the surfactant in the formulation might play a role in increasing the absorption of the drug (Charman et al., 1992).
EVALUATION OF SMEDDS
The primary means of self micro emulsification assessment is visual evaluation. The efficiency of self micro emulsification could be estimated by determining the rate of micro emulsification, droplet size distribution and turbidity measurement.
Droplet size and particle size measurement: The particle size of the micro emulsion is determined by photon correlation spectroscopy or SEM (Scanning Electron Microscopy) which can measure sizes between 10 and 5000 nm. The nanometric size range of the particle is retained even after 100 or 1000 times diluted with distill water, which proves the system’s compatible with excess water (Rad, 2010; Ramachandran et al., 2011).
Refractive index and percent transmission: Refractive index and percent transmittance proves the clearness of formulation. The refractive index of the SMEDDS is measured by refractometer and compared with that of water. The percent transmittance of the system is measured at particular wavelength using UV-Vis spectrophotometer keeping distilled water as blank. If refractive index of system should be similar to that of water. Formulation showing transmittance >99 percent is transparent in nature (Patil et al., 2004; Patil et al., 2007).
Determination of percentage drug content: One capsule of each formulation was taken in a 100 mL volumetric flask, and added 100 mL of extracting solvent. Then mixture was shaken for 1 h in mechanical shaker and kept a side for 24 h. After 24 h, filtered the solution through Whatman filter paper (0.45 μm) to collect the filtrate. The filtrate was then analyzed in UV-spectrophotometer at. The concentration of drug in solution was calculated from absorbance and standard graph (Patil et al., 2007).
Phase separation study: One milliliter SMEDDS was added to glass test tube containing 5 mL of 0.1 N HCl, buffer pH 6.8 and distilled water. After inverting the test tube for 3-4 times, each mixture was stored for a period of 2 h and phase separation was observed visually (Kim and Ku, 2000).
Dispersibility test: The efficiency of self-emulsification of oral micro-/nano-emulsions is measured by using a standard USP dissolution apparatus. To 500 mL of water 1 mL of the formulation is to be added at 37±0.5°C.
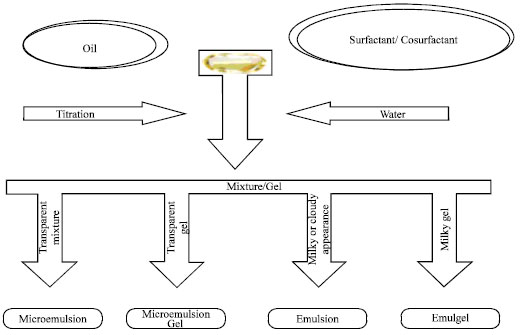 |
| Fig. 4: | Dispersibility test of microemulsion, microgel, emulsion and emulgel in decreasing order of emulsion stability |
A standard stainless steel dissolution paddle rotating at 50 rpm provides gentle agitation. The in vitro performance of the formulations is visually assessed from such a dispersion using a suitable grading system. Grading systems can be based upon the formation of a microemulsion (o/w or w/o), microemulsion gel, emulsion, or emulgel. The schematic flow chart in Fig. 4 illustrates the mode to characterize the type of formulation on the basis of this grading system and the type of dispersion formed on water dilution (Singh et al., 2009b).
Zeta potential measurement: Zeta potential for microemulsion can be determined using a suitable Zetasizer, in triplicate samples (Abbasalipourkabir et al., 2011).
Stability: SMEDDS was diluted with distilled water and to check the temperature stability of samples, they were kept at two different temperature range (2-8°C (refrigerator), room temperature) and observed for any evidences of phase separation, flocculation or drug precipitation.
In order to estimate metastable systems, the optimized SMEDDS formulation was diluted with distilled water. Then microemulsion was centrifuged at 1000 r min-1 for 15 min at 37°C and observed for any alteration in homogeneity of microemulsions (Ghosh et al., 2004).
In vitro release study: In vitro drug release study of SMEDDs formulation was performed by dialysis method, dissolution apparatus 2 and diffusion cell. Study of drug release was done by modified diffusion cell in 200 mL buffer solution 6.8 pH. One gram SMEDDs formulation was placed in boiling tube, both side of boiling tube was opened and one side of tube was tied with cellophane membrane and dipped in buffer solution kept in a beaker below. Upper side of the cylinder was clamped to hold. The beaker was continuously stirred by magnetic stirrer and sample was withdrawn after different time intervals it in straight position and analyzed by UV-Spectrophotometer Percent drug dissolved at different time intervals was calculated using the beer Lambert’s equation (Kang et al., 2004b).
Bioavailability study: Based on the self emulsification properties, particle size data and stability of micro emulsion the formulation is selected for bioavailability studies. The in vivo study is performed to quantify the drug after administration of the formulation. Some of the drugs used in different in vivo models shows in Table 8. The plasma profiles of the drug in experimental animals following oral administration of the conventional tablet and SMEDDS form are compared. Pharmacokinetic parameters of the maximum plasma concentration (Cmax) and the corresponding time (Tmax) for the drug following oral administration are calculated. The area under the concentration-time curve (AUC0→24 h) is estimated according to the linear trapezoidal rule. The relative Bioavailability (BA) of SMEDDS form to the conventional table is calculated using the following Equation Relative BA (%) = (AUC test/AUC reference) X (Dose reference/Dose test).
RECENT ADVANCEMENTS IN SMEDDS
Some of the patents publication on diverse type of lipid formulation shows in Table 9.
Self-emulsifying capsules: After administration of capsules containing conventional liquid SE formulations, micro emulsion droplets form and subsequently disperse in the GI tract to reach sites of absorption. However, if irreversible phase separation of the micro emulsion occurs, an improvement of drug absorption cannot be expected. For handling this problem, sodium dodecyl sulfate was added into the SE formulation (Itoh et al., 2002). With the similar purpose, the super saturable SEDDS was designed, using a small quantity of hydroxyl propyl methyl cellulose (or other polymers) in the formulation to prevent precipitation of the drug by generating and maintaining a supersaturated state in vivo. This system contains a reduced amount of a surfactant, thereby minimizing GI side effect (Gao and Morozowich, 2006; Gao et al., 2003).
| Table 8: | Drug used in different in vivo models |
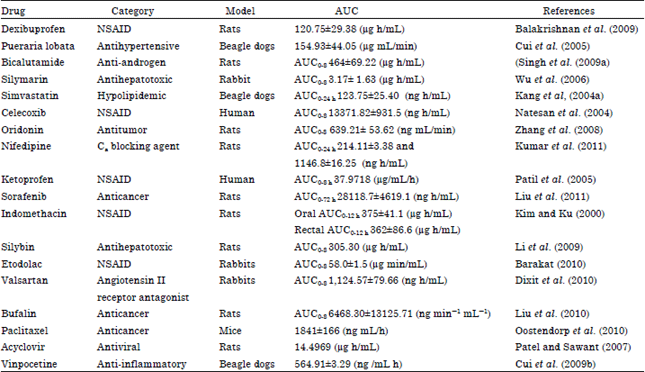 | |
| NSAID- Non Steroidal Anti Inflammatory Drug | |
| Table 9: | Some Patented formulation |
  | |
Self-emulsifying suppositories: Kim and Ku (2000) investigated the Solid-SEDDS could increase not only GI adsorption but also rectal/vaginal adsorption. Glycyrrhizin which by the oral route, barely achieves therapeutic plasma concentrations, can obtain satisfactory therapeutic levels for chronic hepatic diseases by either vaginal or rectal SE suppositories. The formulation included glycyrrhizin and a mixture of a C6-C18 fatty acid glycerol ester and a C6-C18 fatty acid macrogol ester (Takada and Murakami, 2005; Wanwimolruk et al., 1999).
Self-emulsifying nanoparticles: Nanoparticle techniques have been useful in the production of SE nanoparticles. Solvent injection is one of these techniques. In this method, the lipid, surfactant and drugs were melted together and injected drop wise into a stirred non-solvent. The resulting SE nanoparticles were thereafter filtered out and dried. This approach yielded nanoparticles (about 100 nm) with a high drug loading efficiency of 74% (Attama and Nkemnele, 2005). More recently, a novel nanoparticle drug delivery system consisting of chitosan and Glyceryl Monooleate (GMO) for the delivery of Paclitaxel (PTX) has been developed. The SE property of GMO enhanced the solubility of PTX and provided a foundation for chitosan aggregation, meanwhile causing near 100% loading and entrapment efficiencies of PTX (Trickler, 2008).
Self-emulsifying sustained/controlled-release pellets: Formulation of SE controlled-release pellets by incorporating drugs into SES that enhanced their rate of release and then by coating pellets with a water-insoluble polymer that reduced the rate of drug release are also very useful. Pellets were prepared by extrusion/spheronization and contained two water-insoluble model drugs (methyl and propyl parabens); SES contained mono-diglycerides and Polysorbate 80 (Abdalla and Mader, 2007). Figure 5 shows the functioning of the polymer matrix dispersed in a SMEDDS formulation, the composition obtained being in the form of gel capsule. Serratoni et al. (2007) have been developing the combinations of coating and SES could control in vitro drug release by providing a range of release rates and the presence of the SEDDS did not influence the ability of the polymer film to control drug dissolution.
FUTURE PERSPECTIVE
SMEDDS can be an effective solution to the problem of formulating poorly soluble drugs with low solubility in the fluids of the GIT. Although for some time the potential utility of SMEDDS has been known, it is being widely developed and in use only in recent years. The use of a combination of in vitro dispersion and digestion methodologies has enabled a much improved understanding of the role of intestinal lipid processing on the solubilization behavior of lipid based formulations. This in-situ emulsion-forming system with high stability can be taken as an emulsion premix as a formulation. As on date formulation of SMEDDS with drugs having low solubility both in water and in oil is difficult to be developed. With future developments in this novel technology, SMEDDS will remove deficiencies associated with delivery of poorly soluble drugs.
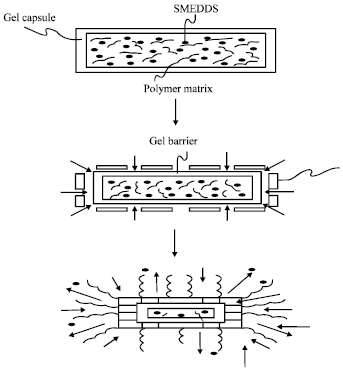 |
| Fig. 5: | Functioning of the polymer matrix dispersed in a SMEDDS formulation |
CONCLUSION
Some of the concealed features of Self micro emulsifying drug delivery systems (SMEDDS) have been revealed by the literature review. SMEDDS is a promising drug delivery system for the enhancement and improvement of bioavailability for a hydrophobic drug. This review article will definitely drag the attention of the young researchers to understand the role of individual lipids and surfactants used for the formulation of SMEDDS as lipid based formulations are still not very widespread as commercial formulations. Also this study explores the possibilities of loading a wide variety of hydrophobic drugs and plant actives as their scale up is convenient as well as economical too.
ACKNOWLEDGMENTS
The authors would like to acknowledge the assistance provided by the Library of Rugnta College of Pharmaceutical Sciences and Research, Kohka Road, Bhilai, Chhattishgarh (India) for collection of literature.
REFERENCES
- Abbasalipourkabir, R., A. Salehzadeh and R. Abdullah, 2011. Cytotoxicity effect of solid lipid nanoparticles on human breast cancer cell lines. Biotechnology, 10: 528-533.
CrossRefDirect Link - Abdalla, A. and K. Mader, 2007. Preparation and characterization of a self emulsifying pellet formulation. Eur. J. Pharm. Biopharm., 66: 220-226.
PubMed - Abdalla, A., S. Klein and K. Mader, 2008. A new self-emulsifying drug delivery system (SEDDS) for poorly soluble drugs: Characterization, dissolution, in vitro digestion and incorporation into solid pellets. Eur. J. Pharm. Sci., 35: 457-464.
CrossRef - Agarwal, V., A. Siddiqui, H. Ali and S. Nazzal, 2009. Dissolution and powder flow characterization of solid self-emulsified drug delivery systeam (SEDDS). Int. J. Pharm., 366: 44-52.
PubMed - Alexander, A., M. Ajazuddin, M. Swarna, M. Sharma and D.K. Tripathi, 2011. Polymers and permeation enhancers: Specialized components of mucoadhesives. Stamford J. Pharm. Sci., 4: 91-95.
CrossRefDirect Link - Amarji, B., Ajazuddin, D. Raghuwanshi, S.P. Vyas and P. Kanaujia, 2007. Lipid Nano Spheres (LNSs) for enhanced oral bioavailability of amphotericin B: Development and characterization. J. Biomed. Nanotechnol., 3: 264-269.
Direct Link - Angare, D., T. Giri, D.K. Tripathi, A. Alexander and Ajazuddin, 2012. Unexplored areas and new findings in lipid emulsion serving as a potential drug carrier for lipophilic drugs: A review. Trends Med. Res., 7: 1-24.
CrossRefDirect Link - Anisa, A.N.I., A.H. Nour and A.H. Nour, 2010. Catastrophic and transitional phase inversion of water-in-oil emulsion for heavy and light crude oil. J. Applied Sci., 10: 3076-3083.
CrossRefDirect Link - Aqil, F., M. Zahin, K.A. El Sayed, I. Ahmad, K.Y. Orabi and J.M. Arif, 2011. Antimicrobial, antioxidant, and antimutagenic activities of selected marine natural products and tobacco cembranoids. Drug Chem. Toxicol., 34: 167-179.
PubMed - Arif, J.M., I. Ahmad and Q. Rahman, 1996. Chrysotile inhibits glutathione-dependent protection against the onset of lipid peroxidation in rat lung microsomes. Pharmacol. Toxicol., 79: 205-210.
PubMed - Atef, E. and A.A. Belmonte, 2008. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Sci., 35: 257-263.
PubMed - Attama, A.A., 2003. The use of solid self-emulsifying systems in the delivery of diclofenac. Int. J. Pharm., 262: 23-28.
PubMed - Bachhav, Y.G. and V. B. Patravale, 2009. SMEDDS of Glyburide: Formulation, in vitro evaluation and stability studies. AAPS Pharm.Sci.Tech., 10: 482-487.
PubMed - Badawi, A.A., M.A. El-Nabarawi, D.A. El-Setouhy and S.A. Alsammit, 2011. Characterization and stability testing of itraconazole solid dispersions containing crystallization inhibitors. Am. J. Drug Discovery Dev., 1: 144-159.
CrossRef - Balakrishnan, P., B.J. Lee, D.H. Oh, J.O. Kim and M.J. Hong et al., 2009. Enhanced oral bioavailability of dexibuprofen by a novel solid Self-Emulsifying Drug Delivery System (SEDDS). Eur. J. Pharm. Biopharm., 72: 539-545.
CrossRefPubMedDirect Link - Barakat, N.S., 2010. Self-emulsifying system for improving drug dissolution and bioavailability: In vitro/in vivo evaluation. Drug Dev. Res., 71: 149-158.
CrossRef - Benet, L.Z. and C.L. Cummins, 2001. The drug efflux‐metabolism alliance: Biochemical aspects. Adv. Drug Deliv. Rev., 50: S3-S11.
CrossRefDirect Link - Borhade, V., H. Nair and D. Hegde, 2008. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS Pharm. Sci. Tech., 9: 13-21.
CrossRefDirect Link - Burcham, D.L., M.B. Maurin, E.A. Hausner and S.M. Huang, 1997. Improved oral bioavailability of the hypocholesterolemic DMP 565 in dogs following oral dosing in oil and glycol solutions. Biopharm. Drug Dispos., 18: 737-742.
Direct Link - Carrigan, P.J. and T.R. Bates, 1973. Biopharmaceutics of drugs administered in lipid-containing dosage forms: GI absorption of griseofulvin from oil in water emulsion in the rat. J. Pharm. Sci., 62: 1476-1479.
CrossRef - Chakraborty, S., D. Shukla, B. Mishra and S. Singh, 2009. Lipid: An emerging platform for oral delivery of drugs with poor bioavailability. Eur. J. Pharm. Biopharm., 73: 1-15.
PubMedDirect Link - Chen, Y., H. Zhang, H. Wang and K. Yang, 2011. Effects of dietary addition of non-ionic surfactants on ruminal metabolism and nutrient digestion of Chinese merino sheep. Asian J. Anim. Vet. Adv., 6: 688-696.
CrossRef - Chitneni, M., K.K. Peh, Y. Darwis, M. Abdulkarim, G.Z. Abdullah and M.J. Qureshi, 2011. Intestinal permeability studies of sulpiride incorporated into Self-Microemulsifying Drug Delivery System (SMEDDS). Pak. J. Pharm. Sci., 24: 113-121.
Direct Link - Cirri, M., P. Mura and P.C. Mora, 2007. Liquid spray formulations of xibornol by using self-microemulsifying drug delivery systems. Int. J. Pharm., 340: 84-91.
Direct Link - Constantinides, P.P., 1995. Lipid microemulsions for improving drug dissolution and oral absorption: Physical and biopharmaceutical aspects. Pharm. Res., 12: 1561-1572.
CrossRefDirect Link - Craig, D.Q.M., S.A. Barker, D. Banning and S.W. Booth, 1995. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int. J. Pharm., 114: 103-110.
CrossRef - Cui, J., B. Yu, Y. Zhao, W. Zhu, H. Li, H. Lou and G. Zhai, 2009. Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems. Int. J. Pharm., 371: 148-155.
CrossRefDirect Link - Cui, S.X., S.F. Nie, L. Li, C.G. Wang, W.S. Pan and J.P. Sun, 2009. Preparation and evaluation of self-microemulsifying drug delivery system containing vinopocetine. Drug Dev. Ind. Pharm., 35: 603-611.
Direct Link - Date, A.A. and M.S. Nagarsenker, 2007. Design and evaluation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for cefpodoxime proxetil. Int. J. Pharm., 329: 166-172.
CrossRefDirect Link - De Gennes, P.G. and C. Taupin, 1982. Microemulsions and the flexibility of oil/water interfaces. J. Phys. Chem., 86: 2294-2304.
CrossRefDirect Link - Devani, M., M. Ashford and D.Q. Craig, 2004. The emulsification and solubilisation properties of polyglycolysed oils in self-emulsifying formulations. J. Pharm. Pharmacol., 56: 307-316.
PubMed - Dey, P., S. Maiti, S. Ray, B. Sa and K. Sen, 2009. Self emulsification of poorly soluble and highly permeable drugs: An overview. Int. J. Pharm. Recent Res., 1: 67-72.
Direct Link - Dintaman, J.M. and J.A. Silverman, 1999. Inhibition of P‐glycoprotein by Dalpha‐tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm. Res., 16: 1550-1556.
PubMed - Gershanik, T. and S. Benita, 1996. Positively charged self-emulsifying oil formulation for improving oral bioavailability of progesterone. Pharm. Dev. Technol., 1: 147-157.
PubMed - Gershanik, T. and S. Benita, 2000. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm., 50: 179-188.
PubMed - Gershanik, T., E. Haltner and S. Benita, 2000. Charge-dependent interaction of self-emulsifying oil formulations with Caco-2 cells monolayers: binding, effects on barrier function and cytotoxicity. Int. J. Pharm., 211: 29-36.
PubMed - Gharaei-Fathabad, E., 2011. Biosurfactants in pharmaceutical industry (A mini-review). Am. J. Drug Discovery Dev., 1: 58-69.
CrossRefDirect Link - Giri, T.K., H. Badwaik, A. Alexander and D.K. Tripathi, 2010. Solubility enhancement of ibuprofen in the presence of hydrophilic polymer and surfactant. Int. J. Applied Biol. Pharm. Technol., 1: 793-800.
Direct Link - Gokturk, S. and U. Var, 2011. Effect of ethanol on partition and binding equilibrium of phenothiazine in anionic and nonionic micellar solutions. Curr. Res. Chem., 3: 49-61.
CrossRefDirect Link - Grove, M., A. Mullertz, J.L. Nielsen and G.P. Pedersen, 2006. Bioavailability of seocalcitol II: Development and characterisation of Self-Microemulsifying Drug Delivery Systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci., 28: 233-242.
PubMed - Gursoy, N.R. and S. Benita, 2004. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophylic drugs. J. Biomed. Pharmacother., 58: 173-182.
PubMedDirect Link - Hauss, D.J., S.E. Fogal, J.V. Ficorilli, C.A. Price, T. Roy, A.A. Jayaraj and J.J. Keirns, 1998. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J. Pharmaceut. Sci., 87: 164-169.
CrossRefPubMedDirect Link - Hoar, T.P. and J.H. Schulman, 1943. Transparent water-in-oil dispersions: The oleopathic hydro- Nature, 152: 102-103.
Direct Link - Hong, J.Y., J.K. Kim, Y.K. Song, J.S. Park and C.K. Kim, 2006. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control Release, 110: 332-338.
Direct Link - Hoque, M.Z., K.M. Hossain and F. Akter, 2009. The effect of lecithin-a non-absorbing emulsifying agent on cookie production. Pak. J. Nutr., 8: 1074-1077.
CrossRefDirect Link - Itoh, K., Y. Tozuka, T. Oguchi and K. Yamamoto, 2002. Improvement of physicochemical properties of N-4472 part I formulation design by using self-microemulsifying system. Int. J. Pharmaceut., 238: 153-160.
CrossRefPubMedDirect Link - Julianto, T., K.H. Yuen and A.M. Noor, 2000. Improved bioavailability of vitamin E with a self emulsifying formulation. Int. J. Pharm., 2: 53-57.
PubMed - Kadu, P.J., S.S. Kushare, D.D. Thacker and S.G. Gattani, 2011. Enhancement of oral bioavailability of atorvastatin calcium by Self-Emulsifying Drug Delivery Systems (SEDDS). Pharm. Dev. Technol., 16: 65-74.
PubMed - Kang, B.K., J.S. Lee, S.K. Chon, S.Y. Jeong and S.H. Yuk et al., 2004. Development of Self-Microemulsifying Drug Delivery Systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharmaceut., 274: 65-73.
CrossRefPubMedDirect Link - Khoo, S.M., A.J. Humberstone, C.J.H. Porter, G.A. Edwards and W.N. Charman, 1998. Formulation design and bioavailability assessment of lipid self emulsifying formulations of halofantrin. Int. J. Pharm., 167: 155-164.
CrossRef - Kim, H.J., K.A. Yoon, M. Hahn, E.S. Park and S.C. Chi, 2000. Preparation and In Vitro evaluation of self-microemulsifying drug delivery systems containing idebenone. Drug Dev. Ind. Pharm., 26: 523-529.
PubMed - Kimura, M., M. Shizuki, K. Miyoshi, T. Sakai, H. Hidaka, H. Takamura and T. Matoba, 1994. Relationship between the molecular structures and emulsification properties of edible oils. Biosci. Biotech. Biochem., 58: 1258-1261.
Direct Link - Kohli, K., S. Chopra, D. Dhar, S. Arora and R.K. Khar, 2010. Self emulsifying drug delivery system: An approach to enhance the oral bioavailability. Drug Discov. Today, 15: 958-965.
PubMed - Lawrence, M.J. and G.D. Rees, 2000. Microemulsion-based media as novel drug delivery systems. Adv. Drug Delivery Rev., 45: 89-121.
CrossRefDirect Link - Li, P., A. Ghosh, R.F. Wagner, S. Krill, M.J. Yatindra and A.T.M. Abu, 2005. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulisons. Int. J. Pharma., 288: 27-34.
PubMed - Li, X.R., Y.S. Pei, Y.Q. Huang, Y.X. Zhou, Y.C. Zhang and Y. Liu, 2009. In vitro and in vivo evaluation of a self microemulsifying drug delivery system for silybin. J. Chin. Pharm. Sci., 18: 342-347.
Direct Link - Liu, Y., P. Zhang, N. Feng, X. Zhang, S. Wu and J. Zhao, 2009. Optimization and in situ intestinal absorption of self-microemulsifying drug delivery system of oridonin. Int. J. Pharm., 365: 136-142.
PubMed - Liu, Y., Z.Q. Chen, X. Zhang, N.P. Feng, J.H. Zhao, S. Wu and R. Tan, 2010. An improved formulation screening and optimization method applied to the development of a self-microemulsifying drug delivery system. Chem. Pharm. Bull., 58: 16-22.
PubMed - Liu, Y.O., J.M. Fan, X.Q. Wang and Q. Zhang, 2011. Preparation of sorafenib self-microemulsifying drug delivery system and its relative bioavailability in rats. J. Chin. Pharm. Sci., 20: 164-170.
Direct Link - Mandal, S. and S.S. Mandal, 2011. Microemulsion drug delivery system: A platform for improving dissolution rate of poorly water soluble drug. Int. J. Pharm. Sci. Nanotechnol., 3: 1214-1219.
Direct Link - Mandawgade, S.D., S. Sharma, S. Pathak and V.B. Patravale, 2008. Development of SMEDDS using natural lipophile: Application to β-Artemether delivery. Int. J. Pharm., 362: 179-183.
CrossRef - Mercuri, A., A. Passalacqua, M.S.J. Wickham, R.M. Faulks, D.Q.M. Craig and S.A. Barker, 2011. The effect of composition and gastric conditions on the self- emulsification process of ibuprofen-loaded self emulsifying drug delivery systems: A microscopic and dynamic gastric model study. Pharm. Res., 28: 1540-1551.
CrossRef - Muherei, M.A. and R. Junin, 2009. Investigating synergism in critical micelle concentration of anionic-nonionic surfactant mixtures: Surface versus interfacial tension techniques. Asian J. Applied Sci., 2: 115-127.
CrossRefDirect Link - Mullertz, A., A. Ogbonna, S. Ren and T. Rades, 2010. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J. Pharm. Pharmacol., 62: 1622-1636.
CrossRef - Natesan, S., R. Subhabrata, K.G. Saroj, B. Ranjan and P.M. Satya, 2004. Formulation design of self-microemulsifying drug delivery systems for improved oral bioavailability of celecoxib. Biol. Pharm. Bull., 27: 1993-1999.
Direct Link - Nekkanti, V.k., P. Karatgi, R. Prabhu and R. Pillai, 2010. Solid Self-microemulsifying formulation for candesartan cilexetil. AAPS Pharm. Sci. Tech., 11: 9-17.
PubMed - Noudeh, G.D., P. Khazaeli and P. Rahmani, 2008. Study of the effects of polyethylene glycol sorbitan esters surfactants group on biological membranes. Int. J. Pharmocol., 4: 27-33.
CrossRefDirect Link - O'Driscoll, C.M. and B.T. Griffin, 2008. Biopharmaceutical challenges associated with drugs with low aqueous solubility- the potential impact of lipid-based formulations. Adv. Drug Deliv. Rev., 60: 617-624.
PubMed - Oostendorp, R.L., T. Buckle, G. Lambert, J.S. Garrigue, J.H. Beijnen, J.H.M. Schellens and O.V. Tellingen, 2011. Paclitaxel in self-micro emulsifying formulations: Oral bioavailability study in mice. Invest New Drugs, 29: 768-776.
CrossRef - Ozawa, K., U. Olsson and H. Kunieda, 1986. Oil-induced structural change in nonionic microemulsions. J. Dispersion Sci. Technol., 22: 119-124.
CrossRefDirect Link - Patel, A.R. and P.R. Vavia, 2007. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J., 9: E344-E352.
CrossRef - Patel, D. and K.K. Sawant, 2007. Oral bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS). Drug Dev. Ind. Pharm., 33: 1318-1326.
PubMed - Patel, D. and K.K. Sawant, 2009. Self micro-emulsifying drug delivery system: Formulation development and biopharmaceutical evaluation of lipophilic drugs. Curre. Drug Delivery, 6: 419-424.
Direct Link - Patel, M.J., N.M. Patel, R.B. Patel and R.P. Patel, 2010. Formulation and evaluation of self-microemulsifying drug delivery system of lovastatin. Asian J. Pharm. Sci., 5: 266-275.
Direct Link - Patel, M., S. Patel, N. Patel and M. Patel, 2011. A review: Novel oral lipid based formulation for poorly soluble drugs. Int. J. Pharm. Sci. Nanotechnol., 3: 1182-1192.
Direct Link - Patil, P. and A. Paradkar, 2006. Porous polystyrene beads as carriers for self-emulsifying system containing loratadine. AAPS Pharm. Sci. Technol., 7: E199-E205.
CrossRef - Patil, P., V. Patil and A. Paradkar, 2007. Formulation of a self-emulsifying system for oral delivery of simvastatin: In vitro and In vivo evaluation. Acta Pharm., 57: 111-122.
Direct Link - Patil, P., P. Joshi and A. Paradkar, 2004. Effect of formulation variables on preparation and evaluation of gelled Self-Emulsifying Drug Delivery System (SEDDS) of ketoprofen. AAPS Pharm. Sci. Tech., 5: 43-50.
CrossRef - Patil, P.R., S. Praveen, R.H.S. Rani and A.R. Paradkar, 2005. Bioavailability assessment of ketoprofen incorporated in gelled self-emulsifying formulation: A technical note. AAPS Pharm. Sci. Tech., 6: E9-E13.
CrossRef - Pogori, N., A. Cheikhyoussef, Y. Xu and D. Wang, 2008. Production and biochemical characterization of an extracellular lipase from Rhizopus chinensis CCTCC M201021. Biotechnology, 7: 710-717.
CrossRefDirect Link - Porter, C.J.H. and W.N. Charman, 2001. In vitro assessment of oral lipid based formulations. Adv. Drug Delivery Rev., 50: S127-S147.
CrossRef - Porter, C.J.H., C.W. Pouton, J.F. Cuine, and W.N. Charman, 2008. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Delivery Rev., 60: 673-691.
CrossRef - Porter, C.J.H., N.L. Trevaskis and W.N. Charman, 2007. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov., 6: 231-248.
CrossRef - Pouton, C.W., 2006. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci., 29: 278-287.
CrossRef - Pouton, C.W., 1997. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev., 25: 47-58.
CrossRef - Rad, A.S., 2010. Study on preparation and some properties of panretin-loaded nanocapsules. Biotechnology, 9: 234-237.
CrossRefDirect Link - Ramachandran, S., G. Thirumurugan and M.D. Dhanaraju, 2011. Development and evaluation of biodegradable chitosan microspheres loaded with ranitidine and cross linked with glutaraldehyde. Am. J. Drug Discovery Dev., 1: 105-120.
CrossRefDirect Link - Reddy, L.H. and R.S. Murthy, 2002. Lymphatic transport orally administered drugs. Indian J. Exp. Boil., 40: 1097-1109.
PubMed - Reiss, H., 1975. Entropy-induced dispersion of bulk liquids. J. Colloids Interface Sci., 53: 61-70.
CrossRef - Seedher, N. and P. Sharma, 2007. Solubility and stability enhancement of poorly-soluble drugs clarithromycin and prednisolone by combination with other drugs. Int. J. Biol. Chem., 1: 229-236.
CrossRefDirect Link - Singh, A.K., A. Chaurasiya, M. Singh, S.C. Upadhyay, R. Mukherjee and R.K. Khar, 2008. Exemestane loaded Self-Microemulsifying Drug Delivery System (SMEDDS): Development and optimization. AAPS Pharm. Sci.Technol., 9: 628-634.
CrossRef - Singh, B., S. Bandopadhyay, R. Kapil, R. Singh and O. Katare, 2009. Self-Emulsifying Drug Delivery Systems (SEDDS): Formulation development, characterization and applications. Crit. Rev. Thera. Drug Carrier Sys., 26: 427-521.
PubMed - Singh, S.K., P.R.P. Verma and B. Razdan, 2009. Development and characterization of a carvedilol loaded self-microemulsifying delivery system. Clin. Res. Reg. Affairs, 26: 50-64.
Direct Link - Tang, J.L., J. Sun and Z.G. He, 2007. Self-emulsifying drug delivery systems: Strategy for improving oral delivery of poorly soluble drugs. Curr. Drug Ther., 2: 85-93.
Direct Link - Tapas, A.R., P.S. Kawtikwar and D.M. Sakarkar, 2011. Modification of felodipine properties using spherically agglomerated solid dispersions. Am. J. Drug Discovery Dev., 1: 160-173.
CrossRefDirect Link - Venkatesh, G., M.I.A. Majid, S.M. Mansor, N.K. Nair, S.L. Croft and V. Navaratnam, 2010. In vitro and in vivo evaluation of self-microemulsifying drug delivery system of buparvaquone. Drug Dev. Indust. Pharmacy, 36: 735-743.
CrossRefPubMedDirect Link - Wanwimolruk, S. and G. Levy, 1987. Effect of age on the pharmacodynamics of phenobarbital and ethanol in rats. J. Pharm. Sci., 76: 503-507.
Direct Link - Wasan, E.K., K. Bartlett, P. Gershkovich, O. Sivak and B. Banno et al., 2009. Development and characterization of oral lipid-based amphotericin B formulations with enhanced drug solubility, stability and antifungal activity in rats infected with Aspergillus fumigatus or Candida albicans. Int. J. Pharm., 372: 76-84.
CrossRefPubMed - Wu, W., Y. Wang and L. Que, 2006. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur. J. Pharm. Biopharma., 63: 288-294.
CrossRefDirect Link - Yadav, O.P., Y.K. Yadav, A.R. Das, T. Dey, S. Kakkar and M.L. Singlav, 2008. Catalytic oxidation of carbonmonoxide using platinum nanoparticles synthesized in microemulsion. Asian J. Scient. Res., 1: 79-84.
CrossRefDirect Link - Zadeh, B.S.M., H. Moghimi, P. Santos, J. Hadgraft, M.E. Lane and F. Rahim, 2010. Formulation of microemulsion systems for improvement of nitrofurazone permeation through silicon membrane as burn wound imitating coverage. Int. J. Pharmacol., 6: 264-270.
CrossRefDirect Link - Zhang, P., Y. Liu, N. Feng and J. Xu, 2008. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int. J. Pharm., 355: 269-276.
CrossRefPubMedDirect Link - Zvonar, A., K. Berginc, A. Kristl and M. Gasperlin, 2010. Microencapsulation of self-microemulsifying system: Improving solubility and permeability of furosemide. Int. J. Pharm., 388: 151-158.
CrossRef - Kim, G.H., J.Y. Lee, Y. Kang, K.N. Kang and E.S. Kim et al., 2011. Preparation and characterization of self-emulsified docetaxel. J. Nanomater.
CrossRef - Porter, C.H.J. and W.N. Charman, 2001. Intestinal lymphatic drug transport: An update. Adv. Drug Delivery Rev., 50: 61-80.
CrossRef - Nazzal, S. and M.A. Khan, 2006. Controlled release of a self-emulsifying formulation from a tablet dosage form: Stability assessment and optimization of some processing parameters. Int. J. Pharm., 315: 110-121.
CrossRefPubMedDirect Link