
Vandana Rai
Human Molecular Genetics Laboratory, Department of Biotechnology, VBS Purvanchal University, Jaunpur-222001 (U.P.), India
Trends in Molecular Sciences
Year: 2011 | Volume: 3 | Issue: 1 | Page No.: 1-13







ABSTRACT
Autism is a neurodevelopmental disability characterized by deficits in verbal communications, impairments in social interactions and repetitive behaviors and recent studies have indicated that autism is considerably more common than previously supposed. Several studies have indicated strong involvement of multigenic components in the etiology of autism. A combination of phenotypic heterogeneity and the likely involvement of multiple interacting loci have hampered efforts at gene discovery. Linkage analyses and candidate gene search approaches so far have not identified any reliable susceptibility genes. This study, will review the literature to date summarizing the results of cytogenetic studies with a focus on recent progress in the field of autism research.
PDF Abstract XML References Citation
Received: May 29, 2010;
Accepted: June 26, 2010;
Published: May 06, 2011
How to cite this article
Vandana Rai, 2011. Autism Genetics and Cytogenetic Abnormalities. Trends in Molecular Sciences, 3: 1-13.
DOI: 10.3923/tms.2011.1.13
URL: https://scialert.net/abstract/?doi=tms.2011.1.13
DOI: 10.3923/tms.2011.1.13
URL: https://scialert.net/abstract/?doi=tms.2011.1.13
INTRODUCTION
Autism (OMIM 209850) is a serious neurodevelopmental disorder characterized by impairments in social interaction, abnormalities in verbal and nonverbal communication and restricted, stereotyped interests and behaviors. The symptoms of autism are discernible in the first 3 years of an affected infant's life and manifest throughout the life span. Approximately 70% of individuals with autistic disorder have some degree of mental retardation and about half are nonverbal or have very impaired speech. Seizures are present by adolescence in about 30% of children with autism and between 5 and 10% of autism cases occur in association with other serious medical conditions such as fragile X, tuberous sclerosis and Angelman’s syndrome (Fombonne, 2003). The prevalence of this disability in the general population is over 1 in 1,000 live births (Fombonne, 2002), although a recent estimate indicate three times higher prevalence rate (Yeargin-Allsopp et al., 2003). Generally, boys are three to four times more commonly affected than girls.
Diagnostically, the autism is highlighted by a triad of symptoms. These include language disturbance, social impairment and a rigid adherence to sameness. There is a spectrum of severity of these symptoms ranging from being almost normal to completely nonfunctioning. The language disturbance ranges from odd to totally nonverbal. The causes of autism are still unknown; however, considerable evidence exists for the involvement of genetic factors. The evidence is based on (1) higher concordance rate among monozygotic compared with dizygotic twins (Cook et al., 1998), (2) strong familial aggregation of autism and (3) sibling recurrence risk is 35 times the population prevalence (Ritvo et al., 1989).
It is now widely accepted that aberrant brain development underlies autism pathogenesis (Rodier et al., 1997; Kemper and Bauman, 2002). Social cognition and communication impairment in autism may be related to dysfunction in amygdale, hippocampus and related limbic and cortical structures. The cerebellum may also form part of a distributed neuronal network responsible for social cognition and communication. Serotonin is the neurotransmitter implicated in autism.
Chromosomal abnormalities detected by cytogenetic assays are of major aid to locate relevant genes for any monogenic and polygenic diseases. A number of such visible breakpoints, translocations, duplications and deletions have been reported for predominantly individual cases of autism spreading over all chromosomes as was extensively reviewed recently (Vorstman et al., 2006). Integration of data from linkage analyses and reports of chromosomal abnormalities are useful to narrow down genomic regions of interest for fine mapping of susceptibility genes.
CYTOGENETIC ABNORMALITIES AND AUTISM CANDIDATE GENES
There are three main approaches to identifying genetic loci, chromosomal regions likely to contain relevant genes: (1) whole genome screens, searching for linkage of autism to shared genetic markers in populations of multiplex families (families with >1 affected family member); (2) cytogenetic studies that may guide molecular studies by pointing to relevant inherited or de novo chromosomal abnormalities in affected individuals and their families; and (3) evaluation of candidate genes known to affect brain development in these significantly linked regions or, alternatively, linkage of candidate genes selected a priori because of their presumptive contribution to the pathogenesis of autism.
Several laboratories around the world are engaged in linkage analysis and positional cloning approaches of genome-wide scans to identify autism susceptibility genes (Ashley-Koch et al., 1999; Barrett et al., 1999; Philippe et al., 1999; Risch et al., 1999; IMGSAC, 1998, 2001; Buxbaum et al., 2001, 2004; Liu et al., 2001; Shao et al., 2002; Alarcon et al., 2005; Auranen et al., 2002, 2005; Yonan et al., 2003; Ylisaukko-Oja et al., 2004; Lamb et al., 2005; McCauley et al., 2005; Ma et al., 2005; Szatmari et al., 2007). These studies have narrowed a number of chromosomal loci and strong involvement of genetic loci on chromosomes 2, 7, 15 and 17 has been indicated.
Genetic evidence from case reports points to a variety of chromosomal abnormalities that are occasional causes of autism. Although 10% of cases of autism are associated with chromosomal abnormalities (Schroer et al., 1998; Folstein and Rosen-Sheidley, 2001) high-resolution cytogenetic scans in families with affected individuals help to locate specific genes or chromosomal regions (loci) potentially associated with the autism. A large number of chromosomal abnormalities have been reported in autism. The study of these abnormalities serves dual purposes. First, these abnormalities may be characteristic of specific sub-syndromes. Second, they point to the chromosomal location of a gene that may have other types of mutations (i.e., point mutations or deletions too small to be visible to karyotyping). In this way, researchers will get hints that a gene or gene system may be involved in patients without visible chromosomal abnormalities.
Because of the frequent association of autism with fragile X syndrome, a genetic linkage between autism and the gene responsible for fragile X (FMR-1) has been investigated in families multiplex for autism. No involvement of the FMR-1 region was found in autistic individuals who had no cytological evidence of fragile X expression. In addition there are some studies which points towards association of fragile sites with autism like 1p31, 3p14, 3q21, 3q27, 6q26, 16q22 (Goldfine et al., 1985), 2q13, 6p23, 12q13 (Arrieta et al., 1996), 1p32, 1q32, 1q44, 2q32, 2q37, 6q21, 7q32, 8q22, 9q32, 10q22, 10q26, 12p12, 14q23, 14q24, 16q23, 16q24, 18q22, 18q23, Xp22 ,Xq22 (Li et al., 1993), 2q31, 2q33, 4q31, 5q21, 5q31, 6p23, 6q26, 12q24, 13q21, Xq22, Xq26 (Manjunatha et al., 2001). Apart from these, common chromosomal polymorphic features like 9h+, long Y, short Y have been reported in autistic children (Gillberg and Wahlstrom, 1985).
Using various stains, the chromosomes of patients with autism are analyzed for visible breakpoints, translocations, duplications and deletions. These regions are then scrutinized for the presence of genes that potentially are involved in the pathogenesis of autism (Vorstman et al., 2006; Christian et al., 2008; Marshall et al., 2008).
 | |
| Fig. 1: | Chromosome 2 showing location of candidate genes |
Some chromosomal regions 2q31-37, 7q21-36, 15q11-q13, 16p11.2, 17q11.2q13 and Xq have gained much attention due to frequent reports of chromosome rearrangements like deletions and/or duplications.
Region 2q31-q37: One region of interest has been the subtelocentric region of 2q31-37 with a higher frequency of deletion in comparison to other chromosomal areas (Marshall et al., 2008). Autism researcher’s interest in subtelomeric region of 2q is generated due to frequent reports of deletions in this region in autistic individuals. This region harbours three genes, DLX1 (2q32), DLX2 (2q32), PAX3 (q35), which are either express in brain or express during neurogenesis (Fig. 1).
The DLX1 (Distal-Less Homeobox 1) and DLX2 genes belong to the DLX family of homeobox transcription factors, which are essential for the development of forebrain GABAergic interneurons during embryonic development. The human DLX2 (distal-less 2) gene has 3 exons and 2 introns (Eisenstat et al., 1999). PAX3 (Paired Box 3) is a DNA-binding protein expressed during early neurogenesis. Two alternatively spliced isoforms are known as PAX3A and PAX3B. PAX3A and PAX3B transcription factors contain 215 and 206 amino acids, respectively. RT-PCR detected high PAX3B expression in esophagus and stomach, with moderate levels in cerebellum, liver and pancreas. PAX3A was expressed only in cerebellum, esophagus and skeletal muscle.
Region 7q21-q36: Several structural rearrangements have been reported in the 7q region in autistic individuals like-dup7p (Wolpert et al., 2000); dup7q31 (Serajee et al., 2003); inv (7) (q22q31.2) (Ashley-Koch et al., 1999); t (7q31) (Vincent et al., 2000); (2;7) (p23;q31.3) (Warburton et al., 2000); t (7;13) (q31.2;q21) (Vincent et al., 1999); t (7;20) (q11.2;q11.2). Chromosomal translocations have also implicated the q22-q33 region of chromosome 7 (Gillberg, 1998; Ashley-Koch et al., 1999; Scherer et al., 2003). The combination of linkage data, the presence of language-related loci and the fact that multiple intriguing candidate genes map to this interval has attracted considerable interest from autism researchers (Rai, 2010).
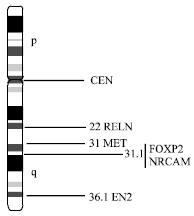 | |
| Fig. 2: | Chromosome 7 shows positions of four autism candidate genes |
This region harbors several genes which are either express in the central nervous system or are related to speech and language functions like- EN2(q36.1), MET (q31), RELN (q22), FOXP2 (q31.1) and NRCAM (q31.1) (Fig. 2).
EN2 (Engrailed 2) gene is located in a chromosomal region that is frequently abnormal in autism (Benayed et al., 2005; Wang et al., 2008). EN2 is a homeobox gene that regulates development of the cerebellum and has attracted attention as a result of the fact that cerebellum abnormalities are among the most consistent findings from pathological and neuroimaging studies in autism. MET is also a strong functional candidate for involvement in autism because it encodes a receptor tyrosine kinsae involved in neuronal growth and organization, as well as immunological and gastrointestinal functioning; these are the systems in which abnormalities have been reported in autism (Campbell et al., 2006). RELN (Reelin) gene encodes a protein that controls intracellular interactions involved in neuronal migration and positioning in brain development (Campbell et al., 2006). The gene reelin (RELN), which localizes to a site of chromosomal translocation at 7q22, is a large secreted glycoprotein potentially involved in neural migration during development (Hong et al., 2000). It is of particular interest given that it binds to neuronal receptors and that the pathology of autism can include migration cell defects (Bailey et al., 1998). Alterations in RELN protein affect cortical and cerebellar development and the cerebellar neuronal abnormalities are among the more robust pathologic findings in autism. Both family-based and population based association studies also indicate that variations in RELN may confer risk to autism (Fatemi et al., 2001; Skaar et al., 2005; Serajee et al., 2006). NRCAM expresses at highest levels in brain, adrenal medulla and adrenal cortex and at intermediate levels in placenta, pancreas, thyroid and testis. This is a surface membrane protein with multiple Ig domains at their N termini followed by several fibronectin type III repeats found in nervous system. FOXP2 (Forkhead Box P2) is a putative transcription factor containing a polyglutamine tract and a forkhead DNA binding domain. It directly regulates expression of the CNTNAP2 gene, encoding a neurexin expressed in developing human cortex, by binding to a regulatory sequence in intron 1. Both FOXP2 and CNTNAP2 are involved in developmental speech and language disorders. Disorders of language and communication are a core feature of the autistic phenotype. The involvement of FOXP2 gene to autism is disputed (Newbury et al., 2002), the several subsequent finding of a breakpoint in the FOXP2 gene in a patient with autism is an important result confirming the presumed involvement of FOXP2.
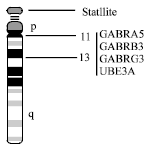 | |
| Fig. 3: | Chromosome 15 showing positions of autism candidate genes |
Region 15q11-q13: Chromosome 15q11-13 is the most frequent site of autosomal abnormalities in autism. These abnormalities most commonly involve duplication, typically as either interstitial duplication or inverted duplicated isodicentric marker chromosomes (Schinzel et al., 1990; Gillberg et al., 1991; Browne et al., 1997; Wolpert et al., 2001), deletion and translocation etc. Deletions of 15q11-q13, though less frequent, have also been reported in autistic individuals, with the deleted material usually of paternal origin (Kerbeshian et al., 1990; Sabrey and Farag, 1998). Several reports are already available about chromosomal abnormalities in this region like-del 15q12 (Kerbeshian et al., 1990; Sabrey and Farag, 1998); dup 15q11-q13 (Baker et al., 1994; Bundey et al., 1994; Flejter et al., 1996; Reppetto et al., 1998; Bolton et al., 2001); maternally derived dup (15) (Robinson et al., 1993; Gurrieri et al., 1999); inv dup (15) (Cook et al., 1997); isodicentric chr (15) (Rineer et al., 1998); triplication (15q11-13) (Hotpof and Bolton, 1995); t (15p;16p) (Martin et al., 2000). Duplications of chromosome 15q11-13 are the commonly recurrent cytogenetic aberration associated with autism and occur in up to 5% of patients with autism (Vorstmann et al., 2006). The cytogenetic abnormalities of chromosome 15q11-q13 point to few gene targets for candidate gene study. This region contains genes like-gamma amino butyric acid (GABAA) receptor genes (15q11.1-q12)-GABRA5, GABRB3, GABRG3 and ubiquitin-protein ligase e3a (UBE3A) (15q11-q13) (Fig. 3).
The gamma-amino butyric acid (GABAA) receptor gene cluster (which contains genes for three of the receptor’s subunits: GABRB3, GABRA5 and GABRG3) is strongly implicated in the pathogenesis of autism, given its involvement in the inhibition of excitatory neural pathways and its expression in early development (Owens and Kriegstein, 2002). GABA is the major inhibitory neurotransmitter in the mammalian CNS and acts by binding to the GABA-A receptor. Any malfunction of these genes may have implications for the inhibition of excitory neural pathways as well as during early brain development and therefore pathological for autism. The most common positive linkage findings were within GABRB3 gene (Martin et al., 2000; Curran et al., 2005; Ashley-Koch et al., 2006).
The apparent gender specificity of the 15q11-q13 abnormalities is presumably attributable to imprinting, an epigenetic mechanism by which only one of a gene’s two inherited alleles is expressed, with expression determined by the allele’s gender of origin (Constancia et al., 1998). In brain tissue (but not elsewhere), UBE3A, the Angellman syndrome gene, is expressed predominantly from the maternally derived allele. Disrupted expression of the maternal UBE3A, therefore produces AS, whereas disruption of the paternally derived allele produces no discernible abnormal phenotype (Knoll et al., 1993). Prader-Willi syndrome gene, conversely, are paternally expressed; the predominant cause of PWS, therefore, is disrupted expression of the paternal copy of the Small Nuclear Ribonucleoprotein Polypeptide N (SNRPN) gene and other contiguous genes.
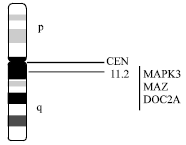 | |
| Fig. 4: | Chromosomes 6 showing positions of autism candidate genes |
UBE3A belongs to a family of functionally related proteins defined by a conserved C-terminal 350-amino acid HECT domain. HECT E3 proteins appear to be important in substrate recognition and in ubiquitin transfer. Rougeulle et al. (1998) showed that imprinting of the UBE3A gene is restricted to brain. Its expression is biallelic in fibroblasts, lymphoblasts, heart, kidney and other tissues.
Region 16p11.2: Microdeletion (Ballif et al., 2007; Ghebranious et al., 2007; Kumar et al., 2008) and duplication (Engelen et al., 2002; Weiss et al., 2008) at 16p11.2 are frequently reported by several investigators in autistic individuals. This small region contains three genes which are express in either in nervous system or during the development of brain - MAPK3, MAZ and DOC2A (Fig. 4).
MAPK3 (Mitogen-Activated Protein Kinase 3) gene is expressed in human and fetal brains. Mapk3-/- mice display abnormal avoidance behavior, hyperactivity and reduced long term potentiation and immune system abnormalities (Mazzucchelli et al., 2002; Kumar et al., 2008). MAPK3 is member of a family of tyrosyl-phosphorylated and Mitogen-Activated Protein Kinases (MAPKs) that participates in cell cycle progression. Several reports concluded that MAPK pathway is necessary for experience-dependent plasticity and for long-term potentiation of synaptic transmission in the developing visual cortex. MAZ (Myc-Associated Zinc Finger Protein) is expressed in several tissues with the highest expression in brain especially in midfrontal cortex. MAZ directly regulates genes involved in GABA signaling, neuronal differentiation and serotonin pathway (Okamoto et al., 2002; Kumar et al., 2008). MAZ gene encodes a transcription factor with dual roles in transcription initiation and termination. DOC2A (Double C2-Like Domain-Containing Protein, Alpha) is expressed predominantly in the brain, localize to synaptic vesicles and is hypothesized to regulate synaptic activity through Ca-dependent mechanisms, consistent with the proposed role of Ca2+ signaling in autism. Doc2a-/- mice display alterations in synaptic transmission and long term potentiation as well as learning and behavioural deficits that include abnormal passive avoidance behavior (Sakaguchi et al., 1999; Kumar et al., 2008).
Region 17q11-q22: Several cytogenetic abnormalities like deletion, inversion are reported in this region in individual with autism like-microdeletion 17q21.31 (Koolen et al., 2006); del 17 (p11.2 p11.3) (Vostanis et al., 1994).
Several autism researchers are interested in this region because it contains gene SLC6A4 (Solute Carrier Family 6 (5-HTT)), the serotonin transporter and during chromosomal rearrangement this gene is impaired in autistic individuals. This may be a candidate gene because of hyperserotonemia observed in approximately 25% of patients with autism (Klauck et al., 1997; Yirmiya et al., 2001; Betancur et al., 2002).
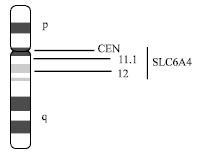 | |
| Fig. 5: | Chromosomes 17 showing positions of autism candidate genes |
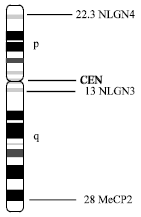 | |
| Fig. 6: | Chromosome X showing positions of autism candidate genes |
SLC6A4 gene lies at 17q11-q12 (Fig. 5). Serotonin (5-hydroxytryptamine; (5-HT) is a neurotransmitter in the central and peripheral nervous systems. Following release, 5-HT is actively cleared from synaptic spaces by SLC6A4, a high-affinity, Na (+)- and Cl (-)-dependent transporter localized in presynaptic neuronal membranes. Serotonin transporter mediates reuptake of serotonin from the synapses. Interest in this gene and its protein products derives from a plausible role for serotonin in the repetitive behaviors observed frequently in patients. Impaired function of serotonin system may result in depression, epilepsy, obsessive-compulsive behavior and affective disorders.
Regions Xp22.3 and Xq13-q28: Hints from the deletion in X chromosome in three autistic females lead to screening of X chromosome for autism candidate genes. The sex chromosomes show abnormalities in several cytogenetic analyses of autistic individuals like- t(X; 8) (p22.13; q22.1) (Bolton et al., 1995); delXp (James et al., 1998; Thomas et al., 1999), sex monosomy and trisomy (Donnelly et al., 2000). Chromosome X harbors three plausible autism candidate genes-NLGN3 (Xq 13), NLGN4 (Xp22.3) and MECP2 (Xq28) (Fig. 6).
NLGN3 (Neuroligins3) gene is located at X q13 whereas NLGN4 (Neuroligin 4) gene lies within the region (Xp22.3) where deletions have been identified in several patients with autism. Coding region mutations in NLGN4 appear to be an uncommon cause of autism and X-linked mental retardation, since hundreds of other subjects have been screened without identifying additional mutations. The nuroligins are cell adhesion proteins with important function in synaptogenesis during brain development and in connection of pre- and post-synaptic membranes. MECP2 (Methyl-CpG-binding protein (2) is a transcriptional repressor that binds to methylated CpG dinucleotides generally located at gene promoters and recruits HDAC1 and other proteins involved in chromatin repression. De novo mutations of the MeCP2 gene located on chromosome Xq28 occur in 80% of female patients with Rett syndrome, a pervasive developmental disorder generally characterized by regression, autism, microcephaly, stereotyped behaviors, epilepsy and breathing problems, whereas in males mutations are generally lethal (Amir et al., 1999). Lam et al. (2000) identified mutations in the MECP2 gene in sporadic cases of autism, whereas no mutations in the MECP2 gene were found in a sample of 59 autistic individuals by Vourch et al. (2001).
CONCLUSION
Research suggests that microscopic cytogenetic abnormalities may contribute to autism approximately in 10% of cases and large number of chromosomal abnormalities have been reported in autism. From research standpoint, chromosomal abnormalities offer an avenue for the rapid identification of candidate regions for gene discovery. This is particularly the case of balanced translocation and chromosomal inversions in which two discrete breakpoints interrupt the normal chromosomal architecture. Likewise, small deletions may point to chromosomal intervals that warrant further study. The value of these types of findings has been demonstrated repeatedly with respect to developmental disorders. Few years back chromosomal abnormalities have led to identification of the NLGN (neuroligin) family genes NLGN3 and NLGN4 as strong candidates for involvement in developmental delay and autism.
REFERENCES
- Amir, R.E., I.B. van den Veyver, M. Wan, C.Q. Tran, U. Francke and H.Y. Zoghbi, 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CPG-binding protein 2. Nat. Genet., 23: 185-188.
CrossRefPubMedDirect Link - Alarcon, M., A.L. Yonan, T.C. Gilliam, R.M. Cantor and D.H. Geschwind et al., 2005. Quantitative genome scan and ordered-subsets analysis of autism endophenotypes support language QTLs. Mol. Psychiatry, 10: 747-757.
PubMed - Arrieta, I., T. Nunez, A. Gil, P. Flores, E. Usobiagu and B. Martinez, 1996. Autosomal folate sensitive sites in an autistic Basque sample. Ann. Genet., 39: 69-74.
PubMed - Ashley-Koch, A., C.M. Wolpert, M.M. Menold, L. Zaeem and S. Basu et al., 1999. Genetic studies of autistic disorder and chromosome 7. Genomics, 61: 227-236.
PubMed - Ashley-Koch, A.E., H. Mei, J. Jaworski, D.Q. Ma and M.D. Ritchie et al., 2006. An analysis paradigm for investigating multi-locus effects in complex disease: Examination of three GABA receptor subunit genes on 15q11-q13 as risk factors for autistic disorder. Ann. Hum. Genet., 70: 281-292.
PubMed - Auranen, M., R. Vanhala, T. Varilo, K. Ayers and E. Kempas et al., 2002. A genomewide screen for autism-spectrum disorders: Evidence for a major susceptibility locus on chromosome 3q25-27. Am. J. Hum. Genet., 71: 777-790.
Direct Link - Bailey, A., P. Luthert and A. Dean, B. Harding and I. Janota et al., 1998. A clinicopathological study of autism. Brain, 121: 889-905.
Direct Link - Baker, P., J. Piven, S. Schwartz and S. Patil, 1994. Brief report: Duplication of chromosome 15q11-13 in two individuals with autistic disorder. J. Autism Dev. Disorder, 24: 529-535.
CrossRef - Ballif, B.C., S.A. Hornor, E. Jenkin, S. Madan-Khetarpa and U. Surti et al., 2007. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet., 39: 1071-1073.
CrossRef - Barrett, S., J.C. Beck, R. Bernier, E. Bisson and T.A. Braun et al., 1999. An autosomal genomic screen for autism. Collaborative linkage study of autism. Am. J. Med. Genet., 88: 609-615.
PubMed - Benayed. R., N. Gharani, I. Rossman, V. Mancuso and G. Lazar et al., 2005. Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am. J. Hum. Genet., 77: 851-868.
PubMed - Betancur, C., M. Corbex, C. Spielewoy, A. Philippe and J.L. Laplanche et al., 2002. Serotonin transporter gene polymorphisms and hyperserotonemia in autistic disorder. Mol. Psychiatry, 7: 67-71.
PubMed - Bolton, P., J. Powell, M. Rutter, V. Buckle, J.R. Yates, Y. Ishikawa-Brush and A.P. Monaco, 1995. Autism, mental retardation, multiple exostoses and short stature in a female with 46,X, t (X;8)(p22.13; q22.1). Psychiatr. Genet., 5: 51-55.
PubMed - Bolton, P.F., N.R. Dennis, C.E. Browne, N.S. Thomas, M.W. Veltman, R.J. Thompson and P. Jacobs, 2001. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Med. Genet., 5: 675-685.
PubMed - Browne, C.E., N.R. Dennis, E. Maher, F.L. Long, J.C. Nicholson, J. Sillibourne and J.C. Barber, 1997. Inherited interstitial duplications of proximal 15q: Genotype-phenotype correlations. Am. J. Hum. Genet., 61: 1342-1352.
PubMed - Bundey, S., C. Hardy, S. Vickers, M.W. Kilpatrick and J.A. Corbett, 1994. Duplication of 15q11-13 region in a patient with autism,epilepsy and ataxia. Dev. Med. Child Neurol., 36: 736-742.
PubMed - Buxbaum, J.D., J.M. Silverman, C.J. Smith, M. Kilifarski and J. Reichert et al., 2001. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am. J. Hum. Genet., 68: 1514-1520.
Direct Link - Buxbaum, J.D., J. Silverman, M. Keddach, C.J. Smith, E. Hollander, N. Ramoz and J.G. Reichert, 2004. Linkage analysis for autism in a subset families with obsessive-compulsive behaviors: evidence for an autism susceptibility gene on chromosome 1 and further support for susceptibility genes on chromosome 6 and 19. Mol. Psychiatry, 9: 144-150.
PubMed - Campbell, D.B., J.S. Sutcliffe, P.J. Ebert, R. Militerni and C. Bravaccio et al., 2006. A genetic variant that disrupts MET transcription is associated with autism. Proc. Natl. Acad. Sci. USA., 103: 16834-16839.
CrossRef - Christian, S.L., C.W. Brune, J. Sudi, R.A. Kumar and S. Liu et al., 2008. Novel Submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry, 63: 1111-1117.
PubMed - Constancia, M., B. Pickard, G. Kelsey and W. Reik, 1998. Imprinting mechanisms. Genome Res., 8: 881-900.
Direct Link - Cook, E.H.J.r, R. Courchesne, C. Lord, N.J. Cox and S. Yan et al., 1997. Evidence of linkage between the serotonin transporter and autistic disorder. Mol. Psychiatry, 2: 247-250.
PubMed - Cook, E.H.Jr., R.Y. Courchesne, N.J. Cox, C. Lord and D. Gonen et al., 1998. Linkage disequilibrium mapping of autistic disorder with 15q11-13 markers. Am. J. Hum. Genet., 62: 1077-1083.
PubMed - Curran, S., S. Roberts, S. Thomas, M. Veltman and J. Browne et al., 2005. An association analysis of microsatellite markers across the Prader-Willi/Angelman critical region on chromosome 15 (q11-13) and autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr Genet., 137: 25-28.
PubMed - Donnelly, S.L., C.M. Wolpert, M.M. Menold, M.P. Bass and J.R. Gilbert et al., 2000. Female with autistic disorder and monosomy X (Turner syndrome): Parent-of-origin effect of the X chromosome. Am. J. Med. Genet., 96: 312-316.
PubMed - Eisenstat, D.D., J.K. Liu, M. Mione, W. Zhong and G. Yu et al., 1999. DLX-1, DLX-2 and DLX-5 expression define distinct stages of basal forebrain differentiation. J. Comp. Neurol., 414: 217-237.
PubMed - Engelen, J.J., C.E. de Die-Smulders, R. Dirckx, W.M. Verhoeven, S. Tuinier, L.M. Curfs and A.J. Hamers, 2002. Duplication of chromosome region 16p) (p11.2->p12.1) in a mother and daughter with mild mental retardation. Am. J. Med. Genet., 109: 149-153.
PubMed - Fatemi, S.H., J.M. Stary, A.R. Halt and G.R. Realmuto, 2001. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J. Autism. Dev. Disorder, 31: 529-535.
PubMed - Flejter, E.W.L., P.E. Bennett-Baker, M. Ghaziuddin, M. McDonald, S. Sheldon and J.L. Gorski, 1996. Cytogenetic and molecular analysis of inv dup(15) chromosome observed in two patients with autistic disorder and mental retardation. Am. J. Med. Genet., 61: 182-187.
PubMed - Folstein, S.E. and B. Rosen-Sheidley, 2001. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet., 2: 943-955.
PubMed - Ghebranious, N., P.F. Giampietro, F.P. Wesbrook and S.H. Rezkalla, 2007. A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder and mild mental retardation. Am. J. Med. Genet., 143: 1462-1471.
PubMed - Gillberg, C. and J. Wahlstrom, 1985. Chromosomal abnormalities in infantile autism and other childhood psychosis: A population study of 66 cases. Dev. Med. Child Neurol., 27: 293-304.
PubMed - Gillberg, C., S. Steffenberg and H. Schaumann, 1991. Autism epidemiology: Is autism more common now than 10 years?. Br. J. Psychiatry, 158: 403-409.
Direct Link - Gurrieri, F., A. Battaglia, L. Torrisi, R. Tancredi, C. Cavallaro, E. Sangiorgi and G. Neri, 1999. Pervasive developmental disorder and epilepsy due to maternally derived duplication of 15q11-q13. Neurology, 52: 1694-1697.
PubMedDirect Link - Hong, S.E., Y.Y. Shugart, D.T. Huang, S.A. Shahwan and P.E. Grant et al., 2000. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet., 26: 93-96.
PubMed - IMGSAC, 1998. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Hum. Mol. Genet., 7: 571-578.
Direct Link - James, R.S., B. Coppin, P. Dalton, N.R. Donnis and C. Mitchell et al., 1998. A study of females with deletions of the short arm of the X chromosme. Hum. Genet., 102: 507-516.
Direct Link - Kerbeshian, J., L. Burd, T. Ranall, J. Martsolf and S. Jalal, 1990. Autism ,profound mental retardation and atypical bipolar in a 33 year old female with a deletion of 15q12(pt 2). J. Ment. Defic. Res., 34: 205-210.
PubMed - Koolen, D.A., L.E. Vissers, R. Pfundt, N. de Leeuw and S.J. Knight et al., 2006. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet., 38: 999-10001.
PubMed - Kumar, R.A., S. KaraMohamed, J. Sudi, D.F. Conrad and C. Brune et al., 2008. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet., 17: 628-638.
PubMed - Lamb, J.A., G. Barnby, E. Bonora, E. Bacchelli and F. Blasi et al., 2005. Analysis of IMGSAC autism susceptibility loci: evidence for sex limited and parent of origin specific effects. J. Med. Genet., 42: 132-137.
CrossRef - Liu, J., D.R. Nyholt, P. Magnussen, E. Parano and P. Pavone et al., 2001. A genome-wide screen for autism susceptibility loci. Am. J. Hum. Genet., 69: 327-340.
PubMed - Ma, D.Q., P.L. Whitehead, M.M. Menold, E.R. Martin and A.E. Ashley-Koch et al., 2005. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am. J. Hum. Genet., 77: 377-388.
PubMed - Manjunatha, K.R., G.K. Chetan, R. Arathi, S. Padma and H.N. Venkatesh et al., 2001. Genetics of Autism: Association of chromosomal fragile sites. IJHG, 1: 293-299.
Direct Link - Martin, E.R., M.M. Menold, C.M. Wolpert, M.P. Bass and S.L. Donnelly et al., 2000. Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am. J. Med. Genet., 96: 43-48.
PubMed - Marshall, C.R., A. Noor, J.B. Vincent, A.C. Lionel and L. Feuk et al., 2008. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet., 82: 477-488.
PubMed - McCauley, J.L., C. Li, L. Jiang, L.M. Olson and G. Crockett et al., 2005. Genome-wide and ordered-subset linkage analyses provide support for autism loci on 17q and 19p with evidence of phenotypic and interlocus genetic correlates. BMC Med. Genet., 6: 1-1.
PubMed - Newbury, D.F., E. Bonora, J.A. Lamb, S.E. Fisher and C.S. Lai et al., 2002. FOXP2 is not a major susceptibility gene for autism or specific language impairment. Am. J. Hum. Genet., 70: 1318-1327.
PubMed - Philippe, A., M. Martinez, M. Guilloud-Bataille, C. Gillberg and M. Rastam et al., 1999. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum. Mol. Genet., 8: 805-812.
Direct Link - Reppetto, G.M., L.M. White, P.J. Bader, D. Johnson and J.H. Knoll, 1998. Interstitial duplications of chromosme region 15q11q13: Clinical and molecular characterization. Am. J. Med. Genet., 79: 82-89.
PubMed - Risch, N., D. Spiker, L. Lotspeich, N. Nouri and D. Hinds et al., 1999. A genomic screen of autism: Evidence for a multilocus etiology. Am. J. Hum. Genet., 65: 493-507.
PubMed - Ritvo, E.R., L.B. Jorde, A. Mason-Brothers, B.J. Freeman and C. Pingree et al., 1989. The UCLA-University of Utah epidemiologic survey of autism: recurrence risk estimates and genetic counseling. Am. J. Psychiatry, 146: 1032-1036.
Direct Link - Rougeulle, C., C. Cardoso, M. Fontes, L. Colleaux and M. Lalande, 1998. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet., 19: 15-16.
PubMed - Sakaguchi, G., T. Manabe, K. Kobayashi, S. Orita and T. Sasaki et al., 1999. Doc2alpha in an activity-dependent modulator of excitatory synaptic transmission. Eur. J. Neursci., 11: 4262-4268.
PubMed - Scherer, S.W., J. Cheung, J.R. MacDonald, L.R. Osborne and K. Nakabayashi et al., 2003. Human chromosome 7: DNA sequence and biology. Science, 300: 767-772.
PubMed - Schinzel, A.A., L. Brecevi, F. Bernasconi, F. Binkert, F. Berthet, A. Wuilloud and W.P. Robinson, 1994. Intrachromosomal triplication of 15q11-q13. J. Med. Genet., 31: 798-803.
CrossRef - Schroer, R.J., M.C. Phelan, R.C. Michaelis, E.C. Crawford and S.A. Skinner et al., 1998. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet., 76: 327-336.
Direct Link - Serajee, F.J., H. Zhong, R. Nabi, A.H. Huq, 2003. The metabotropic glutamate receptor 8 gene at 7q31; partial duplication and possible association with autism. J. Med. Genet., 40: e42-e42.
CrossRef - Shao, Y., C.M. Wolpert, K.L. Raiford, M.M. Menold and S.L. Donnelly et al., 2002. Genomic screen and follow-up analysis for autistic disorder. Am. J. Med. Genet., 114: 99-105.
PubMed - Skaar, D.A., Y. Shao, J.L. Haines, J.E. Stenger and J. Jaworski et al., 2005. Analysis of the RELN gene as a genetic risk factor for autism. Mol. Psychiatry, 10: 563-571.
PubMed - Szatmari, P., A.D. Paterson, L. Zwaigenbaum, W. Roberts, J. Brian and X.Q. Liu, 2007. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet., 39: 319-328.
CrossRef - Thomas, N.S., A.J. Sharp, C.E. Browne, D. Skuse, C. Hardie and N.R. Dennis, 1999. Xp deletions associated with autism in three females. Hum. Genet., 104: 43-48.
PubMed - Vincent, J.B., J.A. Herbrick, H.M. Gurling, P.F. Bolton, W. Roberts and S.W. Scherer, 2000. Identification of a novel gene on chromosome 7q31 that is interrupted by a translocation breakpoint in an autistic individual. Am. J. Hum. Genet., 67: 510-514.
PubMed - Vostanis, P., R. Harrington, M. Prendergast and P. Frandon, 1994. Case reports of autism with interstitial deletion of chromosome 17(p11.2 p11.2) and monosomy of chromosome 5(5pter->5p15.3). Psychiatry Genet., 4: 109-111.
PubMed - Vourch, P., T. Bienvenu, C. Beldjord, J. Chelly, C. Barthelemy, J.P. Muh and C. Andres, 2001. No mutations in the coding region of the Rett syndrome gene MeCP2 in 59 autistic patients. Eur. J. Hum. Genet., 9: 556-558.
PubMed - Wang, L., M. Jia, W. Yue, F. Tang and M. Qu et al., 2008. Association of the ENGRAILED 2(EN2) gene with autism in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatry Genet., 147B: 434-438.
PubMed - Warburton, P., G. Baird, W. Chen, K. Morris, B.W. Jacobs, S. Hodgson and Z. Docherty, 2000. Support for linkage of autism and specific language impairement to 7q3 from two chromosome rearrangements involving band 7q31. Am. J. Med., 96: 228-234.
PubMed - Wolpert, C.M., M.M. Menold, M.P. Bass, M.B. Qumsiyeh and S.L. Donnelly et al., 2000. Three probands with autistic disorder and isodicentric chromosome 15. Am. J. Med. Genet., 96: 365-372.
PubMed - Wolpert, C.M., S.L. Donnelly, M.L. Cuccaro, D.J. Hedges and C.P. Poole et al., 2001. De novo partial duplication of chromosome 7p in a male with autistic disorder. Am. J. Med. Genet., 105: 222-225.
PubMed - Yeargin-Allsopp, M., C. Rice, T. Karapurkar, N. Doernberg, C. Boyle and C. Murphy, 2003. Prevalence of autism in a US metropolitan area. JAMA, 289: 49-55.
PubMed - Yirmiya, N., T. Pilowsky, L. Nemanov, S. Arbelle, T. Feinsilver, I. Fried and R.P. Ebstein, 2001. Evidence for an association with the serotonin transporter promoter region polymorphism and autism. Am. J. Med. Genet., 105: 381-386.
PubMed - Ylisaukko-Oja, T., T.N. von Wendt, E. Kempas, S. Sarenius and T. Varilo et al., 2004. Genome-wide scan for loci of Asperger syndrome. Mol. Psychiatry, 9: 161-168.
PubMed - Yonan, A.L., M. Alarcon, R. Cheng, P.K. Magnusson and S.J. Spence et al., 2003. A genome-wide screen of 345 families for autism-susceptibility loci. Am. J. Hum. Genet., 73: 886-897.
PubMed