
Fazlul Huq
School of Biomedical Sciences, Faculty of Health Sciences, The University of Sydney, Australia
Journal of Pharmacology and Toxicology
Year: 2007 | Volume: 2 | Issue: 1 | Page No.: 63-71







ABSTRACT
Phenytoin (PHT, also known as Dilantin) is a broad-spectrum anticonvulsant that is widely used for the prevention and treatment of seizure disorders. However, it provokes skin rash in 5 to 10% patients and has been found to be teratogenic in various experimental animal species. Epidemiological and clinical studies indicate that women who have taken PHT during pregnancy have an increased risk of bearing a child with a congenital anomaly. The toxic side effects of PHT may result from its primary and secondary metabolites, rather than the parent drug. PHT is metabolised by cytochrome P450 enzymes (CYP2C9 and CYP2C19) primarily to inactive metabolite 5-(4-hydroxyphenyl)-5-phenylhydantoin (4HPPH, in both R- and S-forms, accounting for about 80% of all metabolites). 4HPPH may be further metabolized to catechol that spontaneously oxidizes to semiquinone and quinone species that bind covalently with proteins. Other minor metabolites in man are: 5-(3-hydroxyphenyl)-5-phenylhydantoin (3HPPH) and 5-(3,4-dihydroxy-1, 5-cyclohexadiene-1-yl)-5-phenylhydantoin (DHD). Molecular modelling analyses show that PHT and most of its metabolites do not differ widely in their kinetic lability except Q and SQ which have much lower values. Q has the lowest HOMO-LUMO energy difference and is therefore considered to be most toxic. The differences in heats of formation suggest that Q may be thermodynamically unstable as well that may be subject to both electrophilic and nucleophilic attack.
PDF Abstract XML References Citation
How to cite this article
Fazlul Huq, 2007. Molecular Modelling Analysis of the Metabolism of Phenytoin. Journal of Pharmacology and Toxicology, 2: 63-71.
DOI: 10.3923/jpt.2007.63.71
URL: https://scialert.net/abstract/?doi=jpt.2007.63.71
DOI: 10.3923/jpt.2007.63.71
URL: https://scialert.net/abstract/?doi=jpt.2007.63.71
INTRODUCTION
Phenytoin (5,5-diphenylhydantoin, PHT, also known as Dilantin) is a broad-spectrum anticonvulsant (Liu and Wells, 1995) that is widely used for the prevention and treatment of seizure disorders. However, it provokes skin rash in 5 to 10% patients (Cuttle et al., 2000) and has been found to be teratogenic in various experimental animal species (Harbison 1978; McClain and Langhoff, 1980). It can also produce fever and drug-induced hepatitis (Kinobe et al., 2005) and is known to cause gingival overgrowth as a chronic side effect (Soga et al., 2004). PHT toxicity may result from intentional overdose, dosage adjustments, drug interactions and alterations in physiology (Simon, 2005). Epidemiological and clinical studies indicate that women who have taken PHT during pregnancy have an increased risk of bearing a child with a congenital anomaly (Hanson and Smith, 1975; Buchler et al., 1990). Cerebral neurotoxicity of PHT in mature experimental animals is also well known (Kiefer et al., 1989; Volk et al., 1986). The toxic side effects of PHT may result from its primary and secondary metabolites, rather than the parent drug (Cuttle et al., 2000). PHT has a narrow therapeutic window of 10-20 μg mL-1 in serum (Hara et al., 1999).
PHT produces stabilizing effects on neuronal plasma membrane function, including the ability to block sodium channels, inhibit calcium influx and in certain types of epithelial cells (e.g., glia and choroid plexus) to increase basal membrane sodium permeability.
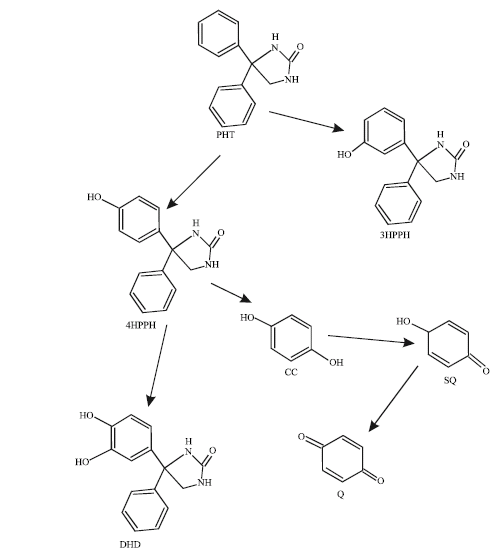 | |
| Fig. 1: | Proposed metabolic pathways for PHT and its metabolites (Hara et al., 1999) |
PHT is metabolised by cytochrome P450 enzymes (CYP2C9 and CYP2C19) primarily to inactive metabolite 5-(4-hydroxyphenyl)-5-phenylhydantoin (4HPPH, in both R- and S-forms, accounting for about 80% of all metabolites). 4HPPH may be further metabolized to catechol that spontaneously oxidizes to semiquinone and quinone species that bind covalently with proteins (Cuttle and Munns et al., 2000). Other minor metabolites in man are: 5-(3-hydroxyphenyl)-5-phenylhydantoin (3HPPH) and 5-(3,4-dihydroxy-1,5-cyclohexadiene-1-yl)-5-phenylhydantoin (DHD) (Hassell et al., 1984). Figure 1 gives the metabolic pathways for PHT.
In this study, molecular modelling analyses have been carried out using the program Spartan ’02 Spartan (2002) to investigate the relative stability of PHT and its metabolites with the aim of providing a better understanding on the relative toxicity due to PHT and its metabolites.
Computation Methods
The geometries of PHT and its metabolites 3HPPH, 4HPPH, SQ, Q and DHD have been optimised based on molecular mechanics, semi-empirical and DFT calculations, using the molecular modelling program Spartan ’02. Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried using the program Spartan ’02 at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.00 was set as the terminating condition. The order of calculations: Molecular mechanics followed by semi-empirical followed by DFT minimized the chances of the structures being trapped in local minima rather reaching global minima. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structures corresponding to global minimum or close to that was reached in most cases. Although RMS gradient of 0.001 may not be sufficiently small for vibrational analysis, it is believed to be sufficiently low for calculations associated with electronic energy levels. For the optimised structures, single point calculations were carried to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The study was carried out in the School of Biomedical Sciences, The University of Sydney during the period September 2005 to March 2006.
RESULTS AND DISCUSSION
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, dipole moment and energies of HOMO and LUMO as per both PM3 and DFT calculations for PHT and its metabolites 3HPPH, 4HPPH, CC, SQ, Q and DHD. Figs. 2-8 give the optimised structures of PHT and its metabolites 3HPPH, 4HPPH, CC, SQ, Q and DHD as per DFT calculations. The structures also give regions of negative electrostatic potential (greyish-white envelopes), HOMOs (blue and red where red indicates HOMOs with high electron density) in (a) and surface charges in (b) where red indicates negative, blue indicates positive and green indicates neutral.
The calculated solvation energies of PHT and its metabolites 3HPPH, 4HPPH, CC, SQ, Q and DHD from PM3 calculations in kcal mol-1 are, respectively -10.12, -14.16, -14.84, -10.34, -8.60,-5.74 and -18.57 and their dipole moments from DFT calculations are 2.68, 1.64, 3.85, 0.00, 2.04, 0.00 and 2.65, respectively. The range of solvation energy values indicate that PHT and its metabolites would differ in their solubility in water. DHD is expected to be most soluble in water followed by 4HPPH and 3HPPH whereas Q is expected to be least soluble in water. The higher solubility of DHD as compared to PHT is due to the presence of two polar hydroxyl groups in DHD and that with respect to CC is due to presence of the hydantoin ring.
In the case of PHT, 3HPPH, 4HPPH and DHD, the electrostatic potential is found to be more negative around the carbonyl oxygen atom and a nitrogen atom of the hydantoin ring, indicating that the positions may be subject to electrophilic attack. In the case of 3HPPH, 4HPPH, DHD, CC and SQ, the electrostatic potential is also found to be more negative around the hydroxyl oxygen atoms, indicating that the positions may be subject to electrophilic attack. In the case of SQ and Q, the electrostatic potential is also found to be more negative around the carbonyl oxygen atoms, indicating that the positions may be subject to electrophilic attack.
| Table 1: | Calculated thermodynamic and other parameters of phenytoin and its metabolites |
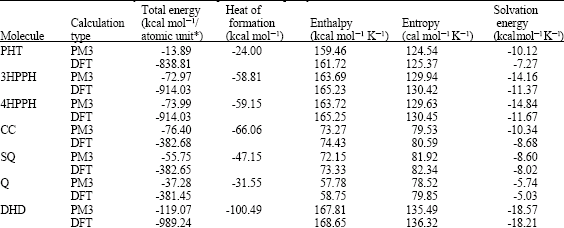 | |
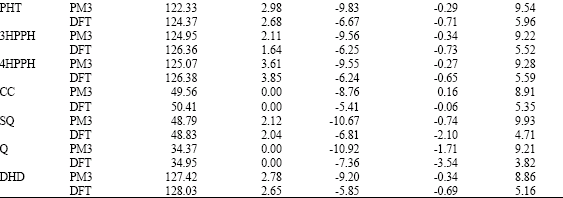 | |
| * in atomic unit from DFT calculations | |
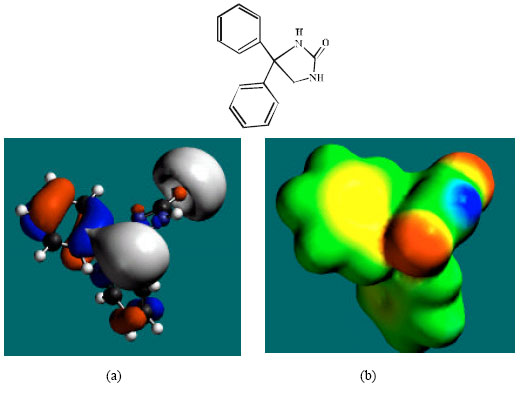 | |
| Fig. 2: | Structure of PHT giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
The surface of PHT is found to abound in green and yellow regions and also contains two red lobes and a blue one, indicating the interaction of PHT with can be amphiphilic i.e., partly hydrophobic and partly hydrophilic so that PHT would have moderate solubility in both water and lipids. As compared to PHT, the surface of 3HPPH and 4HPPH abound less in green, more in yellow, red and blue lobes, indicating the interaction of 3HPPH and 4HPPH with biomolecules including water would be more hydrophilic than that of PHT so that 3HPPH and 4HPPH would be more soluble in water than PHT. It may be noted that a similar conclusion was reached earlier from solvation energy values. The surface of CC is found to abound more in red and yellow regions than green regions, indicating the interaction of CC with biomolecules is more likely to be hydrophilic than hydrophobic so that CC would be soluble in water.
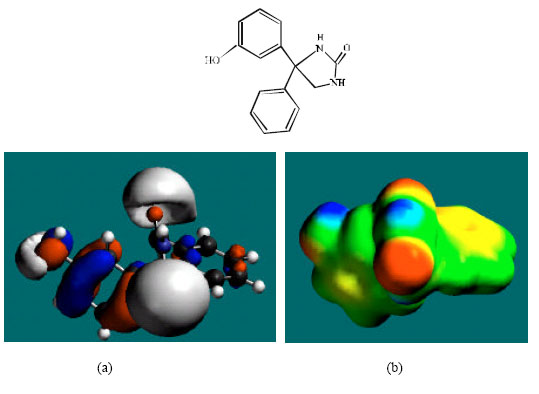 | |
| Fig. 3: | Structure of 3HPPH giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
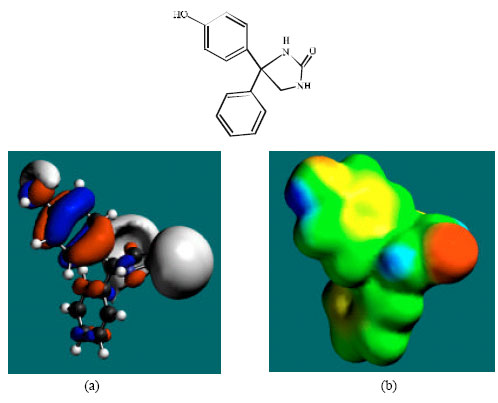 | |
| Fig. 4: | Structure of 4HPPH giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
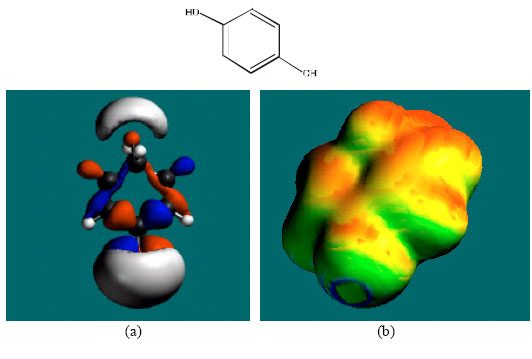 | |
| Fig. 5: | Structure of CC giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
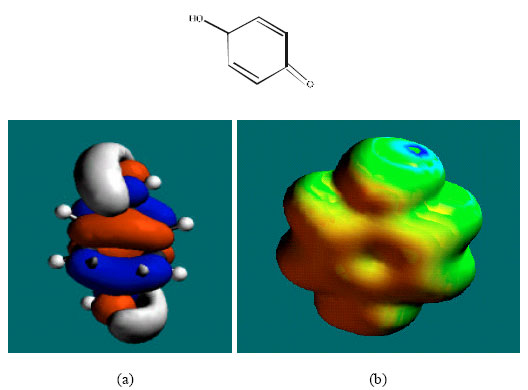 | |
| Fig. 6: | Structure of SQ giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
The surface of SQ is found to abound more in green than red, yellow or blue regions, indicating the interaction of SQ and Q with biomolecules is more likely to be more hydrophobic than hydrophilic so that SQ would be expected to be more soluble in lipid than in water.
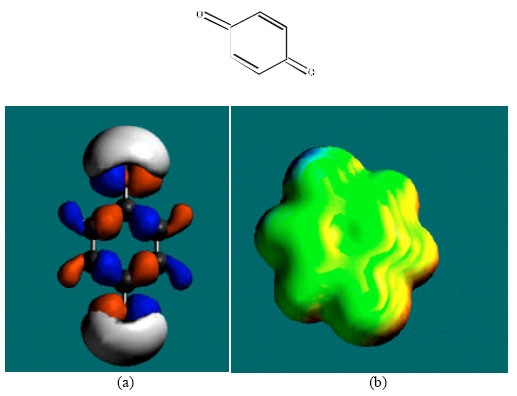 | |
| Fig. 7: | Structure of Q giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
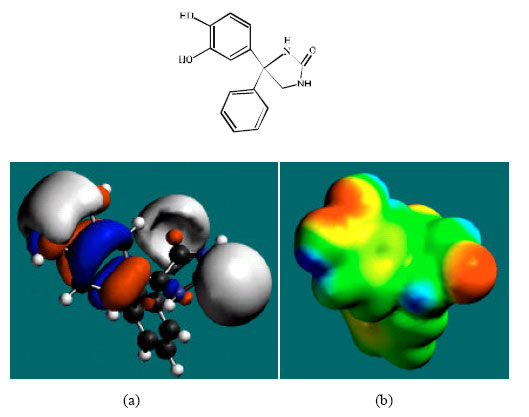 | |
| Fig. 8: | Structure of DHD giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral |
The surface of DHD is found to abound in green, red, blue and yellow regions, indicating the interaction of DHD with biomolecules is more likely to be hydrophilic than hydrophobic so that DHD would be expected to be more soluble in water than in lipid.
The HOMOs with high electron density are found above and below some or all of the phenyl ring carbon atoms in the case of PHT, 3HPPH, 4HPPH, CC, SQ and Q. Other positions around which the HOMOs with high electron density are found include atoms of hydantoin ring. The overlap or closeness of positions of negative electrostatic potential and HOMOs with high electron density at some positions e.g. the hydroxyl group of 3HPPH and 4HPPH reinforce the idea that the positions may be subject to electrophilic attack.
Placed in the order of increasing HOMO-LUMO energy differences (from highest to lowest), PHT and its metabolites are: PHT>4HPPH>3HPPH>CC>DHD>SQ>Q, indicating that PHT would be least labile kinetically and Q most labile. Since for xenobiotics kinetic lability rather than thermodynamic stability is expected to be a more important determinant of toxicity (as biochemical reactions may be coupled so that an otherwise improbable reaction may occur spontaneously), Q is considered to be most toxic and PHT is considered to be least toxic. Q may also be thermodynamically unstable as it has a higher heat of formation that SQ and CC. It was noted earlier that Q is a toxic molecule that binds spontaneously with proteins.
CONCLUSION
Molecular modelling analyses show that PHT and most of its metabolites do not differ widely in their kinetic lability except Q and SQ which have much lower values. Q has the lowest HOMO-LUMO energy difference and is therefore considered to be most toxic. The differences in heats of formation suggest that Q may be thermodynamically unstable as well that may be subject to both electrophilic and nucleophilic attack.
ACKNOWLEDGMENTS
Fazlul Huq is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Buchler, B.A., D. Delimont, M. van Waes and R.H. Finnell, 1990. Prenatal prediction of risk of the fetal hydantoin syndrome. N. Engl. J. Med., 322: 1567-1572.
Direct Link - Cuttle, L., A.J. Munns, N.A. Hogg, J.R. Scott, W.D. Hooper, R.G. Dickson and E.M.J. Gillam, 2000. Pheytoin metabolism by human cytochrome P450: Involvement of P450 3A and 2C forms in secondary metabolism and drug-protein adduct formation. Drug. Metabol. Dispos., 28: 945-950.
Direct Link - Hara, S., J. Hagiwara, M. Fukuzawa, N. Ono and T. Kuroda, 1999. Determination of phenytoin and its metabolites in human serum by high-performance liquid chromatography with fluorescence detection. Anal. Sci., 15: 371-375.
Direct Link - Kinobe, R.T., O.T. Parkinson, D.J. Mitchell and E.M.J. Gillam, 2005. P450 2C18 catalyzes the metabolic bioactivation of phenytoin. Chem. Res. Toxicol., 18: 1868-1875.
Direct Link - Liu, L. and P.G. Wells, 1995. Potential molecular targets mediating chemical teratogenesis: In vitro peroxidase-catalyzed phenytoin metabolism and oxidative damage to proteins and lipids in murine maternal hepatic microsomes and embryonic 9000g supernatant. Toxicol. Applied Pharmacol., 134: 71-80.
Direct Link - Soga, Y., F. Nishimura, Y. Ohtsuka, H. Araki and Y. Iwamoto et al., 2004. CYP2C polymorphisms, phenytoin metabolism and gingival overgrowth in epileptic subjects. Life Sci., 74: 827-834.
CrossRefDirect Link