
Fazlul Huq
Faculty of Health Sciences, C42, School of Biomedical Sciences, The University of Sydney, Lidcombe, NSW, Australia
Journal of Pharmacology and Toxicology
Year: 2007 | Volume: 2 | Issue: 3 | Page No.: 295-299







ABSTRACT
Meropenem (MER) is a new carbapenum antibiotic that is highly active in the treatment of a broad range of pathogenic infections including gram-positive and gram-negative bacteria. It is water-soluble and eliminated mainly by renal excretion, through both glomerular filtration and tubular secretion. MER is metabolized into open ring metabolite UK-1a which is also microbiologically active. In healthy volunteers, 70% of the administered dose is excreted as the unchanged drug and 20% as the metabolite UK-1a, in the urine. There is a significant reduction in renal excretory capacity for MER and its metabolite UK-1a in elderly subjects. Molecular modelling analyses based on molecular mechanics, semi-empirical and DFT calculations show that both MER and UK-1a have large LUMO-HOMO energy differences so that they would be kinetically inert. Also, neither MER nor UK-1a is found to abound in electron-deficient regions so that they would not readily react with glutathione and nucleobases in DNA. This may explain why the side-effects from MER-therapy are low.
PDF Abstract XML References Citation
How to cite this article
Fazlul Huq, 2007. Molecular Modelling Analysis of the Metabolism of Meropenem. Journal of Pharmacology and Toxicology, 2: 295-299.
DOI: 10.3923/jpt.2007.295.299
URL: https://scialert.net/abstract/?doi=jpt.2007.295.299
DOI: 10.3923/jpt.2007.295.299
URL: https://scialert.net/abstract/?doi=jpt.2007.295.299
INTRODUCTION
Meropenem (MER; ICI 194,660; (4R,5S,6S)-3-[(3S,5S)-5-(dimethylcarbamoyl)-3-pyrrlidinylthio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3,2,0]hept-2-ene-2-carboxylic acid) is a new carbapenum antibiotic that is highly active in the treatment of a broad range of pathogenic infections including gram-positive and gram-negative bacteria (Back et al., 1989). It shows good tolerability at high doses and a low incidence of seizures that makes it particularly useful in treating serious infections (Robatel et al., 2002). MER is water-soluble and eliminated mainly by renal excretion, through both glomerular filtration and tubular secretion (Nilsson-Ehle et al., 1991). The drug is more stable than imipenem against renal dehydropeptidase-1 so that it is not necessary to administer any renal dehydropeptidase-1 inhibitor in order to achieve high concentrations in urine. It shows good tolerability at high doses and produces low incidence of seizures that makes it particularly suitable for the treatment of serious infections (Robatel et al., 2002).
MER is metabolized into open ring metabolite UK-1a which is also microbiologically active. In healthy volunteers, 70% of the administered dose is excreted as the unchanged drug and 20% as the metabolite UK-1a, in the urine Burman et al., 1991. There is a significant reduction in renal excretory capacity for MER and its metabolite UK-1a in elderly subjects (Ljungberg and Nilsson-Ehle, 1992).
In this study, molecular modelling analyses have been carried out using the program Spartan, 02 (Spartan 2002) to investigate the relative stability of MER and its metabolite with the aim of providing a better understanding of their relative toxicity. The study was carried out in the School of Biomedical Sciences, The University of Sydney during May to October 2006.
COMPUTATIONAL METHODS
The geometries of MER and its metaboliteUK-1a have been optimised based on molecular mechanics, semi-empirical and DFT calculations, using the molecular modelling program Spartan, 02.
Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimised structures, single point calculations were carried out to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations: molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases. Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
RESULTS AND DISCUSSION
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment and energies of HOMO and LUMO as per both PM3 and DFT calculations for MER and its metabolite UK-1a. Figure 2 and 3 give the regions of negative electrostatic potential (greyish-white envelopes) in (a), HOMOs (where red indicates HOMOs with high electron density) in (b), LUMOs in (c) and density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) in (d) as applied to the optimised structures of MER and UK-1a.
The calculated solvation energies from PM3 calculations of MER and UK-1a are, respectively -15.08 and -13.41 kcal mol-1 and corresponding dipole moments from DFT calculations are 3.0 and 2.7. The values suggest that MER and UK-1a would only be moderately soluble in water.
The LUMO-HOMO energy differences for MER and UK-1a from DFT calculations are 5.41 and 5.51 eV, respectively indicating that both the compounds would be kinetically inert. In the case of MER and UK-1a, the electrostatic potential is found to be more negative around the oxygen, nitrogen and sulfur centres, indicating that the positions may be subject to electrophilic attack.
In the case of MER and UK-1a, both the HOMOs with high electron density and the LUMOs are found to be centred mostly on the dimethylcarbamoyl moiety and the five-membered heterocyclic ring to which it is bonded.
The overlap of HOMO with high electron density and region of negative electrostatic potential close to sulfur, gives further support to the idea that the position may be subject to electrophilic attack.
The molecular surfaces of MER and UK-1a are found to abound in neutral (green) and negative (yellow and red) regions, indicating that the compounds may be subject to lyophilic and electrophilic attacks.
| Table 1: | Calculated thermodynamic and other parameters of MER and its metabolites |
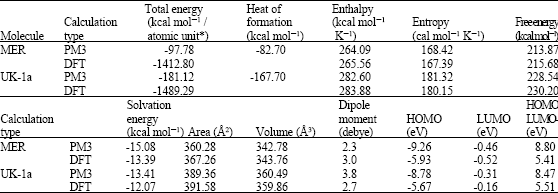 | |
| * in atomic units from DFT calculations | |
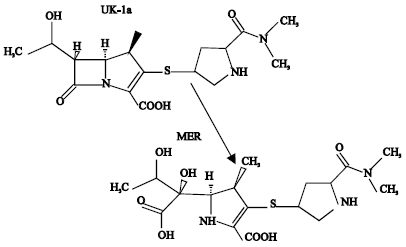 | |
| Fig. 1: | Metabolic pathways for MER (Based on Robatel et al., 2002) |
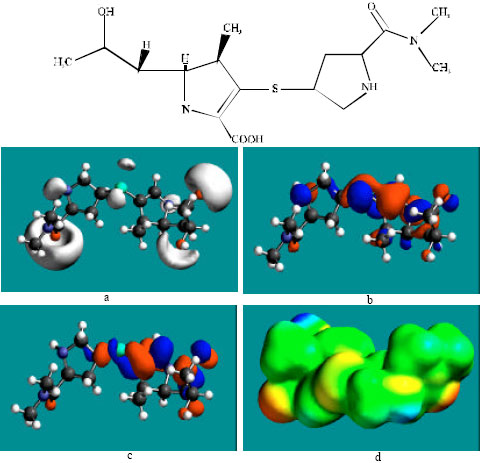 | |
| Fig. 2: | Structure of MER giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density), (c) the LUMOs (where blue indicates LUMOs) and in, (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
Although both the molecular surfaces of both the compounds also possess some electron-deficient (blue) regions so that they may be subject to nucleophilic attacks such as those by glutathione and nucleobases in DNA, in actual fact the rates of such adverse reactions are expected to be low because of the kinetic inertness of the molecules.
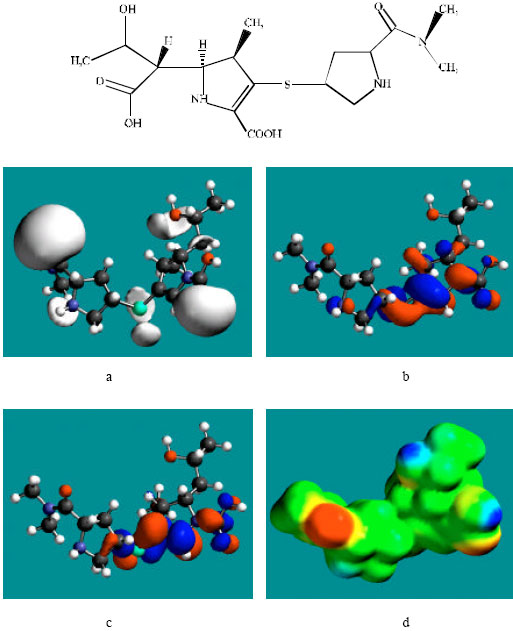 | |
| Fig. 3: | Structure of MER giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density), (c) the LUMOs (where blue indicates LUMOs) and in, (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
When surface area and volume of MER are compared with those of UK-1a, it is found that the values differ to some extent. However, as stated earlier, both MER and UK-1a are found to be pharmacologically active so that both can act as substrate for key enzyme. The results illustrate that the binding between receptor and substrate are more likely to involve induced fit.
CONCLUSION
Molecular modelling analyses based on semi-empirical and DFT calculations show that MER and its metabolite UK-1a have large LUMO-HOMO energy differences so that they would be kinetically inert. This means that although the molecular surfaces of MER and UK-1a abound to possess some electron-deficient regions so that they could react with glutathione and nucleobases in DNA, in actual fact the rate of such adverse reactions may not be significant.
Abbreviations
| MER: | Meropenem; (4R,5S,6S)-3-[[3S,5S)-5-(dimethylcarbamoyl)-3-pyrrlidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3,2,0]hept-2-ene-2-carboxylic acid); ICI 194,660; |
| DFT: | Density functional theory |
| UMO: | Lowest unoccupied molecular orbital |
| HOMO: | Highest occupied molecular orbital |
ACKNOWLEDGMENTS
Fazlul Huq is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Burman, L.A., I. Nilsson-Ehle, M. Hutchinson, S.J. Haworth and S.R. Norrby, 1991. Pharmacokinetics of meropenem and its metabolite ICI 213,689 in healthy subjects with known renal metabolism of imipenem. J. Antimicrob. Chemother., 27: 219-224.
Direct Link - Ljungberg, B. and I. Nillsson-Ehle, 1992. Pharmacokinetics of meropenem and its metabolite in young and elderly healthy men. Antimicrob. Agents Chemother., 36: 1437-1440.
Direct Link - Nilsson-Ehle, I., M. Hutchinson, S.J. Haworth and S.R. Norrby, 1991. Pharmacokinetics of meropenem compared to iminopenem with cilastain in young and healthy males. Eur. J. Clin. Microbiol., 10: 85-88.
Direct Link - Robatel, C., T. Buclin, P. Eckert, M.D. Schaller, J. Biollaz and L.A. Decosterd, 2002. Determination of meropenem in plasma and filtrate-dialysate from patients under continuous veno-venous haemodialfiltration by SPE-LC. J. Pharmaceut Biomed. Anal., 29: 17-33.
Direct Link