
Fazlul Huq
School of Biomedical Sciences,
Faculty of Health Sciences, C42, The University of Sydney,
P.O. Box 170, Lidcombe, NSW 1825,
Australia
Journal of Pharmacology and Toxicology
Year: 2006 | Volume: 1 | Issue: 6 | Page No.: 537-544







ABSTRACT
Maprotiline (also known as Ludiomil) (MAP) is a tetracyclic antidepressant that is chemically and functionally similar to tricyclic antidepressants (TCAs). It is a selective norepinephrine (NE) re-uptake blocker. The side effects of MAP include: nausea, nervousness, increased sweating, cholinergic blockade, cardiac complications such as ventricular tachycardia and atrial fibrillation and allergic reactions. Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that MAP and all its metabolites have large LUMO-HOMO energy differences so that they would be kinetically and therefore less likely to react readily with biomolecules. However, their low solvation energy and hence high lipid solubility suggest that the compounds would have low clearance rate. It is suggested that the toxicity of MAP and its metabolites may be associated with their low clearance rate.
PDF Abstract XML References
How to cite this article
Fazlul Huq, 2006. Molecular Modelling Analysis of the Metabolism of Maprotiline. Journal of Pharmacology and Toxicology, 1: 537-544.
DOI: 10.3923/jpt.2006.537.544
URL: https://scialert.net/abstract/?doi=jpt.2006.537.544
DOI: 10.3923/jpt.2006.537.544
URL: https://scialert.net/abstract/?doi=jpt.2006.537.544
INTRODUCTION
Depression is a common mental disorder that affects more than 120 million people worldwide (Sarah et al., 2005). It is a chronic or recurrent illness that affects both social and economic functions of the sufferers and can lead to suicidal behaviour. According to the World Health Organization mood disorder will be the second leading contributor to the global burden of disease by the year 2020 (Sarah et al., 2005). The most widely accepted cause of depression is believed to be due to dysfunction of either the nonadrenergic or seretonergic neurotransmitter systems (Burrows et al., 1998). Maprotiline (also known as Ludiomil) (MAP) is a tetracyclic antidepressant that is chemically and functionally similar to tricyclic antidepressants (TCAs) (Smith and Reynard, 1992). The side effects of MAP include: nausea, nervousness, increased sweating, cholinergic blockade, cardiac complications such as ventricular tachycardia and atrial fibrillation and allergic reactions. The increased heart rate due to MAP is due its ability to block reuptake of noradrenalin (NE) in the synaptic cleft (Flakovic et al., 1990)
The metabolites of MAP in humans are N-desmethylmaprotiline (NDMAP), N-acetylmaprotiline (AcMAP), N-methylmaprotiline (NMMAP) and N-acetyldesmethylmaprotiline (AcNDMMAP). The drug is metabolized predominantly by cytochrome P450 isoforms CYP2D6 and CYP1A2 with former being the major one (Brachtendorf et al., 2002) although it has been suggested that CYP2C19 isoform may be involved in the metabolism of MAP in patients with major depression who fail to respond to MAP therapy (Vormfelde et al., 1997). Figure 1 summarizes the metabolic pathway of MAP in humans.
In this study, molecular modelling analyses have been carried out using the program Spartan ’02 in the School of Biomedical Sciences, University of Sydney during December 2005-March 2006 to investigate the relative stability of MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP with the aim of providing a better understanding on the relative toxicity of the drug and its metabolites.
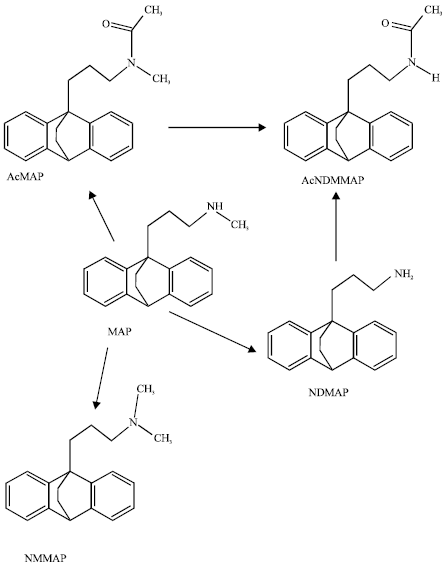 | |
| Fig. 1: | Metabolic pathway for MAP (Sarah et al., 2005; Smith and Reynard, 1992) |
Computational Methods
The geometries of MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP have been optimized based on molecular mechanics, semi-empirical and DFT calculations, using the molecular modelling program Spartan ’02. Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried using the program Spartan ’02 at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimized structures, single point calculations were carried to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations:
Molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases. Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
RESULTS AND DISCUSSION
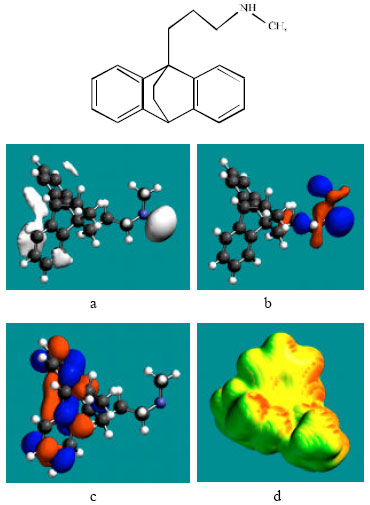
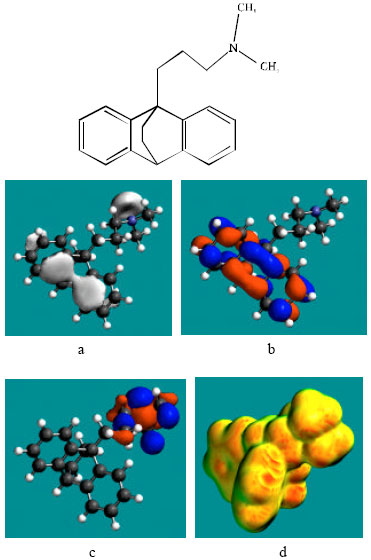
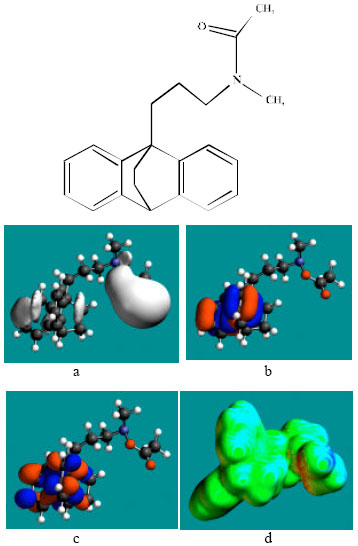
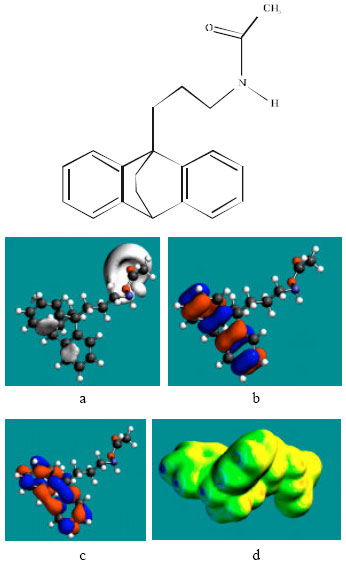
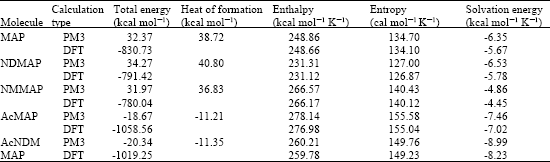
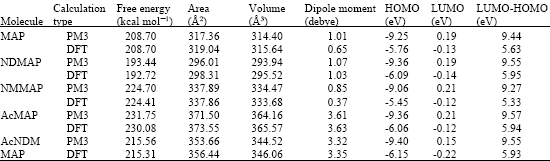
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment, energies of HOMO and LUMO as per both PM3 and DFT calculations for MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP. Figure 2-6 give the regions of negative electrostatic potential (greyish-white envelopes) in (a), HOMOs (where red indicates HOMOs with high electron density) in (b), LUMOs in (c), and surface charges (where red indicates negative, blue indicates positive and green indicates neutral) in (d) as applied to the optimized structures of MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP.
 | |
| Fig. 2: | Structure of MAP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density), (c) the LUMOs (where blue indicates LUMOs) and in (d) surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) |
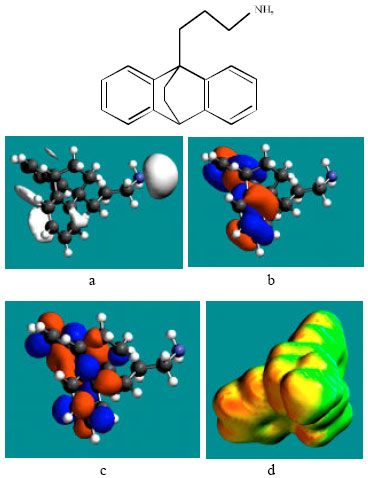 | |
| Fig. 3: | Structure of NDMAP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density), (c) the LUMOs (where blue indicates LUMOs) and in (d) surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) |
The calculated solvation energies of MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP from PM3 calculations in kcal mol-1 are respectively -6.35, -6.53, -4.86, -7.46 and -8.99 and their dipole moments from DFT calculations are 0.65, 1.03, 0.37, 3.63 and 3.35, respectively. Relatively low values for solvation energy and dipole moment suggest that MAP and its metabolites will have low solubility in water and higher solubility in lipids suggesting that the compounds would have low clearance rate which in turn may serve to increase toxicity. Slightly higher solvation energy values found for the metabolites AcMAP and AcNDMMAP are believed to be due to increased molecular size rather than any significant increase in solubility in water. That the surfaces of the two metabolites are found to be less charged than those of MAP, NDMAP and NMMAP (considered later) also support the idea that their interaction with biomolecules is less likely to be hydrophilic.
The HOMO-LUMO energy differences obtained from DFT calculation for MAP and its metabolizes NDMAP, NMMAP, AcMAP and AcNDMMAP are respectively 5.63, 5.95, 5.33, 5.94 and 5.93. These relatively large values indicate that MAP and all its metabolites would be kinetically inert.
 | |
| Fig. 4: | Structure of NMMAP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density), (c) the LUMOs (where blue indicates LUMOs) and in (d) surface electric charges (where red indicates negative, blue indicates positive and green indicates neutral) |
NMMAP would have a slighter greater kinetic lability as it has a lower LUMO-HOMO energy difference. Arranged in the order of increasing kinetic lability MAP and its metabolites are: NMMAP>MAP>AcNDMMAP>AcMAP>NMMAP.
In the case of MAP, NDMAP and NMMAP, the electrostatic potential is found to be more negative around the nitrogen atom of the amino side chain, above and below the two phenyl rings indicating that the positions may be subject to electrophilic attack. In the case of AcMAP and AcNDMMAP, the electrostatic potential is found to be more negative around the nitrogen atom of the acyl group boned to amino nitrogen, above and below the two phenyl rings, again indicating that the positions may be subject to electrophilic attack.
In the case of MAP and NDMAP, the HOMOs with high electron density are found close to the non-hydrogen atoms of the amino side chain and carbon atoms of the cyclohexane ring whereas the LUMOs are found close to the non-hydrogen atoms of the two phenyl rings.
 | |
| Fig. 5: | Structure of AcMAP giving the electrostatic potential and the HOMOs in (a) and surface electric charges in (b) where red indicates negative, blue indicates positive and green indicates neutral) |
In the case of NMMAP and AcNDMMAP, the HOMOs with high electron density are found close to the non-hydrogen atoms of the two phenyl rings whereas the LUMOs are found close to the atoms of the amino side chain. In the case of AcMAP, HOMOs with high electron density and LUMOs are found close to the non-hydrogen atoms of the two phenyl rings. The overlap of HOMO with high electron density and region of negative electrostatic potential close to sulfur, gives further support to the idea that the position may be subject to electrophilic attack.
The greater abundance of red and yellow regions on the surfaces of MAP, NDMAP and NMMAP than that of AcMAP and AcNDMMAP indicate that surfaces of MAP, NDMAP and NMMAP are slightly more negatively charged than those of AcMAP and AcNDMMAP. This means that the interaction of MAP, NDMAP and NMMAP with biomolecules is less likely to be lyophilic than that of AcMAP and AcNDMMAP.
MAP, NDMAP and NMMAP are found to have positive heat of formation values whereas AcMAP and AcNDMMAP have similar negative values. In the absence of calculation of Gibb’s free energy change it is not possible to conclude whether the reactions such as the formation of NDMAP from MAP or the formation of NMMAP from MAP would be spontaneous or not. When surface area and volume of MAP, NDMAP and NMMAP are compared, it is found to that they do not differ widely (Table 1) so that they may be able to associate with the same binding sites taken into account the dynamic nature of enzymes. Thus the metabolites NDMAP and NMMAP may also contribute to the activity of MAP.
 | |
| Fig. 6: | Structure of AcNDMMAP showing distribution of surface charges |
| Table 1: | Calculated thermodynamic and other parameters of MAP and its metabolites |
 | |
 | |
Finally, although the analysis has shown MAP and all its metabolites will have high lipid solubility and low clearance rate (that contribute to their toxicity), MAP and its metabolites are found to be kinetically inert so that they would not react readily with biomolecules.
CONCLUSIONS
Molecular modelling analyses based on semi-empirical and DFT calculations show that. MAP and all its metabolites have large LUMO-HOMO energy differences and hence they would be kinetically inert. This means that none of the molecules may react readily with biomolecules and therefore none of the molecules is expected to be highly toxic. However, all of the compounds are expected to have a low clearance rate because of greater solubility in lipids and this may be a determinant of their toxicity.
ACKNOWLEDGMENT
Fazlul Huq is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.