
I.A. Adejoro
Not Available
T.O. Bamkole
Not Available
Journal of Applied Sciences
Year: 2005 | Volume: 5 | Issue: 9 | Page No.: 1559-1563







ABSTRACT
In this study, using the PM3 Hamiltonian of the quantum mechanical computer code called MOPAC, a procedure we devised for the computation of the Arrhenius parameters of the pyrolysis of some alkyl acetates is reported. The acetates studied are ethyl, n- and I-propyl, I-butyl, t-butyl and s-butyl. The results obtained compare well within an order of magnitude with the experimental values in the literature. It is indeed gratifying that for s-butyl acetate where more than one olefinic product is possible, our procedure reasonably predicts the experimental product ratio in the literature and gives by implication correct Arrhenius parameters for the decomposition of s-butyl acetate in to these separate products.
PDF Abstract XML References Citation
How to cite this article
I.A. Adejoro and T.O. Bamkole, 2005. Semi-empirical Quantum Mechanical, Molecular Orbital Method using Mopac: Calculation of the Arrhenius Parameters for the Pyrolysis of Some Alkyl Acetates. Journal of Applied Sciences, 5: 1559-1563.
DOI: 10.3923/jas.2005.1559.1563
URL: https://scialert.net/abstract/?doi=jas.2005.1559.1563
DOI: 10.3923/jas.2005.1559.1563
URL: https://scialert.net/abstract/?doi=jas.2005.1559.1563
INTRODUCTION
Though literature is replete with reports of systematic experimental study of the kinetics of many classes of reactions in solution and in the gas phase, such report is scanty concerning the quantitative calculation of reaction rates. Theoretical calculation of Arrhenius parameters is attempted here for the gas phase pyrolysis of alkyl acetates, many of which have been studied experimentally[1-6], both for its preparative value and the justification of developments in the theory of reaction rates, to see if the Arrhenius parameters determined experimentally can be obtained by calculation. It has long been known that the basis of such calculation is the Schrodinger wave equation for the electron, which has been successfully employed to give satisfactory qualitative explanations or interpretations of many chemical properties and phenomena. However, its use for the derivation of quantitative data such as Arrhenius parameters of chemical reactions has been hampered for a long time by the mathematical complexity and repetitiveness of the necessary computational procedures, further compounded by electron correlation consideration, even for the simpler, less complicated case of gas phase reactions. However, considerable progress has been made within the last three decades in the development of dependable ab-initio and semi-empirical molecular orbital methods to make possible now, fair attempts to compute reaction rates. Our focus here is essentially to show that the semi-empirical approach can now successfully predict the experimental results recorded in the literature and we here describe our procedure, using the PM3 Hamiltonian in MOPAC[7].
MATERIALS AND METHODS
Geometry definition: The geometry of the reactant is defined in terms of the numbering in structure A below:
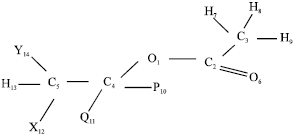 |
Numbering Sequence A
In this structure P, Q, X and Y are substituents, which depend on the substrate being considered. The hydrogen atoms around each C atom in the rough trial geometry were fixed consecutively in the order of the dihedral angles being about 180, -60 and +60 degrees, respectively. H12, H13 and H14 are then β-H atoms that can be eliminated. The usual rules of geometry definition in MOPAC brochure are then applied. This geometry option, coded Option I of numbering sequence A, therefore makes three calculations possible.
If H13 and H14 were interchanged, H13 now has a dihedral angle of about +60 degrees in the trial geometry and by token of making the arrangement of H atoms on C5 different from that of other C atoms which bear hydrogen atoms, becomes the β H atom to be eliminated. This could be referred to as geometry Option II. This would make another calculation possible, making four in all.
Reaction path study: Reaction path studies were performed for each substrate using H13-O6 bond length as the reaction coordinate. This internal coordinate was varied systematically in many small steps from its initial value (say 4.58 A in ethyl acetate) to its value in the product molecule(s) obtained (about 0.9 A in acetic acid and ethylene). All other internal coordinates (bond lengths, bond angles and the dihedral angles) are allowed to optimize throughout the reaction path calculation. As reported by McIver and Kormonicki[8] for other similar work, this resulted in geometries with increasing energy (more positive heats of formation) and at some unpredictable point, the geometry suddenly suffered an abruptly large relaxation to a product-like structure with a concomitant drop in the heat of formation to some value approximately the same as the sum for the expected products (olefin and acetic acid). The product-like structure was then optimized to give the product geometry.
Saddle calculation, locating and refining the transition state geometry: The input data consist of the geometries of both the substrate and product, specified with the same connectivities. The keywords SADDLE and XYZ are invoked. The output of the saddle calculation consists of a progressive alteration and comparison of the geometries and heats of formation of both the reactant and products in a systematic manner to converge at a maximum in heat of formation. At the convergence, we are in the region of the approximate transition state and some gradient minimization procedure [9] (keywords SIGMA or TS) was then used to optimize the resulting approximate transition state geometry and determine its heats of formation.
Characterizing the transition state: A true saddle point is a point on the potential energy surface where a displacement along any direction, except towards the starting material(s) and product(s), will lead to an increase in energy. To characterize the transition state obtained, it was subjected to vibrational analysis, which yields the force constants within the molecule. The presence of five (for a linear system) or six eigenvalues, which are very small and exactly one negative force constant characterizes the system under study as a true transition state.
Intrinsic Reaction Coordinate (IRC) calculations: This is to show that the transition state confirmed by vibrational analysis as a true saddle point actually connects the reactant and the products. It consists of allowing two perturbations to act separately on the transition state (one positive, IRC=1 and the other negative, IRC=-1). One should lead to the geometry of the reactant while the other should lead to the geometry of the product. These IRC calculations are an excellent way of simulating reactions and enable numerous structures along the reaction coordinate to be generated.
Force calculation: This is the same process of vibrational analysis carried out in IRC, but now performed on ground state geometry or system of interest. It yields force constants and the variation of thermodynamic properties with temperature for the species of interest but here there is no negative force constant. It is a prerequisite for thermodynamic calculation. The implementation procedure is as described in the MOPAC brochure[10].
Calculation of rotational barriers: The values of thermodynamic properties calculated from MOPAC are limited to situations in which free internal rotations are absent i.e. for situations in which barriers are so to say, already installed to prevent rotations. This barrier has to be removed. Hence relevant rotational barriers calculated have to be subtracted from the apparent enthalpy of reaction to obtain the true enthalpy. Path studies involving rotations about the relevant bonds were carried out on each reactant: e.g., for ethyl acetate, the O4-C1 and C5-C4 bonds.
Processing of computer output: Although we used the numbering sequence indicated in Structure A above, we may point out that other numbering sequences are possible but output may not be equally easy to process. For example, if we use the numbering sequence B below, the heat of formation using PM3 is –408.64 kJ mol-1 compared to -413.66 kJ mol-1 obtained for sequence A.
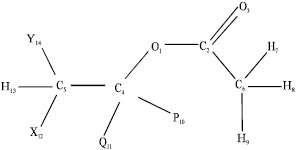 |
Numbering Sequence B
Therefore, for purposes of comparison, we need to use a consistent numbering sequence in the stepwise calculations on a substrate as well as in going from one substrate to another. Moreover, the numbering sequence is important when we consider the relevance of the rotational barriers. For instance, the rotation of the methyl group in the acetyl functional group may appear relevant in sequence B and not in A. Thus, the numbering sequence used may be very important in determining the accuracy of result as well as the ease of obtaining it. Although other sequences are possible, these two were extensively investigated and it was found that A was easier to use.
When the work was begun, it was intended to explore the Hamiltonians present in MOPAC, particularly MINDO/3, MNDO, AM1 and PM3, to find which will correctly predict rates and Arrhenius parameters of acetates. It was soon realized that PM3 was more advanced and had better capabilities so far as present purpose is concerned and so we soon settled to use PM3 solely for our investigation. Finally, where two or more β H atoms are present in a molecule, it would be reasonable to do calculations involving their elimination in turns to see which of them nature will preferentially do business with and so as previously indicated, both option I and option II calculations were done for each substrate.
RESULTS
Ethyl acetate will be used here as an example.
The rotational barriers: The values of some dihedral angles in going from the Ground State (GS) to the Transition State (TS) structure, through etacreac, the reactant structure obtained through IRC calculation (Table 1).
For each rotation, the variation of the heat of formation (ΔH, kJ mol-1) with the relevant dihedral angle (<) is recorded (Table 2) minimum on the first row, maximum on the next; the difference on the third row being the barriers encountered in a 3600 cycle. Using rotation about the C4-O1 bond as an example, i.e. for changes in dihedral angle 5-4-1-2, the following 3-part barrier is obtained.
The interpretation of the above table is that in going from the dihedral angle of -179.160 in the GS through etacreac to 11.770 in the TS, a rotational barrier of 26.35 kJ (comprising the first two of the three-part barrier) is crossed.
In a similar way, rotation about O1-C2 bond produced a 2-part barrier of which none is crossed.
For the rotation of the C4-C5 bond, the dihedral 12-5-4-1 was used and out of the 3-part barrier obtained, only the first two (10.88 kJ) are crossed.
| Table 1: | Variation of dihedral angles (degrees, 0), in going from Ground State (G S) to Transition State (TS) through etacreac |
 | |
| Table 2: | Variation of heat of formation (ΔH kJ mol-1) with the indicated dihedral angle (<, 0), (here 5-4-1-2) |
 | |
The barrier produced by C2-C3 bond rotation (i.e. the rotation of the methyl group on the acetyl functional group) is, (using our option A structure), small and negligible for all the alkyl acetates.
From the use of H13-O6 bond length as the reaction coordinate, only one barrier is encountered and the value is 3.26 kJ.
Therefore, for ethyl acetate, the sum of the relevant barriers (i.e. 26.35 + 10.88 + 3.26 = 40.49 kJ) serves as correction to the apparent activation enthalpy.
The activation energy: The apparent enthalpy of activation is obtained for each substrate through FORCE calculation on both the GS and TS geometries by subtracting the enthalpy of the reactant at 623 K from the enthalpy of the TS at 623 K {i.e. -124.78 – (-363.50) = 238.72 kJ mol-1}. The correction computed is subtracted from the apparent enthalpy of activation for reason already stated. Therefore, for ethyl acetate, the corrected enthalpy of activation is (238.72 – 40.49 = 198.23 kJ mol-1)
According to the Transition State Theory (TST), for a unimolecular reaction
Ea = ΔH + RTwhere ΔH is the corrected enthalpy of activation. Assuming that
T= 623 K then the activation energy Ea = 203.42 kJ mol-1.
Pre-exponential factor, A: For each substrate, the apparent entropy of activation (ΔSapp) is obtained from the FORCE calculation by subtracting the entropy of the substrate at 623 K from the entropy of the transition state also at 623 K. Corrections due to relevant rotational barriers as well as to symmetry considerations are then applied.
The rotational entropy corrections were calculated according to the principle discussed by O’Neal and Benson[11] involving the use of symmetrical co-axial top molecules. This way the entropy of free rotation, Sf, is obtained.
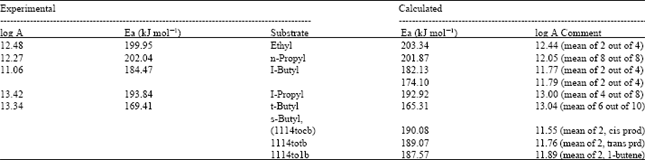
| Table 3: | The calculated as well as the experimental Arrhenius parameters (Ea) kJ mol-1 and A) |
 | |
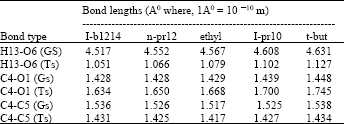
| Table 4: | Variation of bond lengths in the Ground State (GS) and Transition State (TS) for the substrates studied |
 | |
1/n of the Sf value (where n is the number of minima encountered in a 360 degree rotation) is then taken as the contribution of that rotation to the correction for restricted rotation, depending whether the rotation is found relevant or not. As indicated above, the relevance or otherwise of a rotation is determined by whether or not the barrier is encountered and crossed in moving from the Ground State (GS) to the Transition State (TS), movement being in the direction that traverses the geometry of the reactant obtained by Intrinsic Reaction Coordinate (IRC) analysis on the TS.
Besides the corrections for rotation calculated above, corrections (Rlnσ) have to be made for symmetry, where, σ is the number of equivalent β-hydrogen atoms available for elimination. It is otherwise known as the reaction path degeneracy. This is added, not subtracted, as it can only enhance reaction. In this way, the true or corrected entropy of activation was obtained and from it, A was calculated using the equation:
A = (ekT/h) exp {(ΔS/R)} |
since the reaction is unimolecular.
This whole process was carried out for each substrate studied. The results obtained are given in the Table 3 with experimental values for ease of comparison.
DISCUSSION
It is obvious that the experimental and calculated parameters are in good agreement and that the rate constants calculable from the calculated parameters are within an order of magnitude of the experimental ones (Table 3).
Some observations from geometries obtained: In going from the ground state to the transition state geometries, the following remarkable changes in bond lengths are observed: (Table 4) (I) a remarkable decrease in Hx-O6, (ii) a noticeable increase in C4-O1 and (iii) an also noticeable decrease in C4-C5 bonds where, x is either 12, 13 or 14 and other numbers indicate atom-numbers in structure A. These are indicative respectively of the tendency of the β hydrogen on C5 to be abstracted as a proton by O6, the tendency of C4-O1 to elongate and break and the development of double bond character in C4-C5 bond.
We now focus on the transition state geometries. While noting that the effects of α and β methyl substitutions are in the same direction (increase) for the C4-C5 bond, we note a contrasting effect on the H13-O6 and C4-O1 bonds; α methyl substitution lessens the O6-H13 attraction (seen as increasing bond length) while the opposite is noted for β methyl substitution. Similarly, α methyl substitution increases the C4-O1 extension while β methyl substitution does the opposite. While the effect of α and β methyl substitution on C4-C5 bond is non-discriminatory as far mechanistic options are concerned, their effects on H13 – O6 and C4 - O1 are diagnostic of the importance of these bond-length changes in the formation of the transition state. That is, the tendency of the β hydrogen on C5 to be abstracted as a proton by O6 and the tendency of C4-O1 to elongate and break, are very important in the rate-determining step of the reaction. This supports the conclusion of Maccoll[12], based on experimental results that the mechanism for the thermal decomposition of esters involves a synchronous break in the β C-H and α C- O (ether oxygen) bonds through a six-centered transition state driven by heterolysis.
Products from s-butyl acetates: If the reaction of s-butyl acetate to yield cis- and trans-2-butenes and 1-butene is simultaneous and each first order and if the rate constants for production of each product are kcb, ktb and k1b respectively, then overall first order rate constant for the disappearance of s-butyl acetate is (kcb + ktb +k1b) and the overall product ratio should be:
% cis-2-butene = kcb/(kcb + ktb +k1b)
% trans-2-butene = ktb/(kcb + ktb +k1b) and
% 1-butene = k1b/(kcb + ktb +k1b)
From the Arrhenius parameters computed and presented above, this ratio can be easily calculated at any temperature of our choice. Let us choose to calculate for 623 K, i.e. 3500 C, about the middle of the usual temperature range for pyrolytic studies. From the means of the parameters computed, the desired ratio is cis: trans: 1-butene = 15.27: 30.42: 54.31.
Froemsdorf et al.[13] obtained the product ratio of but-1-ene: but-2-ene = 57: 43 at 4500 C. Using a flow system, Emovon and Maccoll[14] obtained the ratio to be 52.4: 47.6. However, using a static system, their result was 56.3: 43.7. Combining this with their analysis which showed the but-2-ene obtained to consist of 64% trans product, their product distribution is cis: trans: but-1-ene = 17.14: 30.46: 52.4 using the flow system and 15.73: 27.97: 56.3, using the static system.
It is concluded that the product distribution computed in this work is in good agreement with these experimental results.
CONCLUSIONS
Therefore, we can see that using the computer code MOPAC and the procedure developed here, we can now obtain quantitative kinetic results for reactions, which are within an order of magnitude of the experimental results. It is also important to highlight that the procedure developed and reported here may have a special fascination for the chemical engineer in the area of reactor design. To the chemical engineer, reactor design[15] is a very important exercise or problem. Among others, it comprises two consecutive steps, viz., (I) the study of the rate at which the chemical reaction of interest occurs and the variables which affect it and (ii) the use of the rate data so obtained to determine how large the equipment he needs should be to obtain the required quantity of product. The first of these steps is the subject of chemical kinetics, here tackled, while the second is the design problem proper. Using our procedure, the chemical engineer can now predict quantitatively reaction rates from the knowledge of the reacting species and possible reaction mechanism and avoid rigorous experimental study of reaction kinetics as a mandatory prelude to the reactor design problem.
REFERENCES
- Maccoll, A., 1958. Gas-phase eliminations. Part I. The unimolecular gas-phase pyrolysis of some esters and analogous compounds. J. Chem. Soc., 1: 3398-3402.
CrossRef
Achor Philemon Okpanachi Reply
I need the comprehensive material on pyrolysis of acetate to study